
摘要:本文探讨了生成式AI在分子输出方面的新颖性与创造性问题,这是一个常被过度兴奋情绪所掩盖的微妙而深刻的挑战。同时,它也直接冲击着药物研发的核心使命:既要确保生成结果具备可受保护的知识产权(IP),更要推动真正具有突破性的治疗创新。
作者:Serhii Vakal. (August 19, 2025) How Novel Are The AI-Generated Molecules? A Reality Check. Available at: https://www.linkedin.com/pulse/how-novel-ai-generated-molecules-reality-check-serhii-vakal-xdguf
欢迎回到我们关于制药研发中生成式AI的系列文章,这一领域既充满挑战又蕴含无限希望。在之前的几篇文章中,我们拨开迷雾,确立了一些来之不易的认知:例如,炫酷的3D分子生成器常常无法通过基础物理验证(第2篇);“可合成性”是不可妥协的前提,而非事后补救的条件(第4篇);而当前最务实的应用方向是增强——而非替代——药物化学家的能力(第5篇)。今天,我们将探讨另一个常被过度宣传所掩盖、却极为微妙且深刻的问题:AI所生成分子的新颖性。
我们常听到这样的说法:AI能够探索高达1060数量级的化学空间——这一数字之庞大,甚至令天文数字都黯然失色——终将突破人类创造力的边界,开启一个全新药物类别的时代。这样的愿景无疑极具吸引力:AI将绘制出真正未知的化学版图,提出精巧的分子骨架,一举解决诸如选择性、活性和ADME等复杂难题。然而,现实远比这更为复杂。身处一线的计算化学家们,肩负着将AI输出转化为实际资产的重任,他们发现:尽管这些工具产能惊人,却常常陷入一种“似曾相识”的困境。系统生成的分子虽然在统计学意义上独一无二,但结构上多为已知化学骨架的衍生变体,往往只是对已有结构进行细微且显而易见的修饰。这绝非学术上的吹毛求疵,而是直接关系到药物研发的核心使命:我们既要确保所生成的分子具备可受保护的知识产权(IP),更要推动真正具有突破性意义的治疗创新。
“新颖”与”有价值”之间的差距
让我们以数据为基础展开讨论,从轶事转向分析。Xie、Zhu和Huang等人最近对71篇已发表的AI设计活性化合物案例研究进行了全面综述,得出了一个令人发人深省的结论。与已知活性化合物相比,人工智能生成的分子中仅有42.3%达到了Tanimoto系数(Tc)低于0.4的水平——这一被广泛认可(甚至可以说是相当宽松)的阈值,通常用于判定分子结构具有合理新颖性。仔细想想:从一开始,从新颖性的角度来看,AI驱动的努力有超过50%的可能性实际上是在重复造轮子。
深入挖掘数据后,我们发现不同AI方法之间存在显著的性能差距,这一差距对研发战略具有重要启示。基于配体的模型表现最为吃力,这类模型直接从已知活性分子列表中学习。它们生成的化合物中近60%与现有化合物高度相似(\(Tc>0.4\))。这并不令人意外——本质上你训练的是一个专业模仿者,其核心功能是识别并复制训练集中定义成功的分子模式。这类模型在生成”看起来像已知药物”的分子方面表现超群,却难以突破创新边界。
相比之下,基于结构的模型从蛋白质靶点的三维几何结构出发,表现明显更优。超过82%的输出结果达到了新颖性阈值(意味着仅有18%的化合物具有高Tc值)。这揭示了一个关键策略要点:你如何要求AI解决问题,从根本上决定了答案的创造力。基于结构的方法更侧重于解决复杂的三维拼图(将新的化学钥匙嵌入特定的生物锁中),而非简单模仿,因此天然能产生更多突破常规的创新解决方案。
也许最能说明问题的是关于真正卓越新颖性的统计数据——那种能开创新药物类别的发现。在所有71项研究中,只有8.4%的分子达到了低于0.2的Tanimoto系数,表明这是一个具有真正新颖性的分子骨架。这才是真正的”现实检验”。AI作为突破性发现的完整方案( turnkey engine)承诺尚未实现。虽然潜力存在,但截至目前,它并不是一个必然结果。
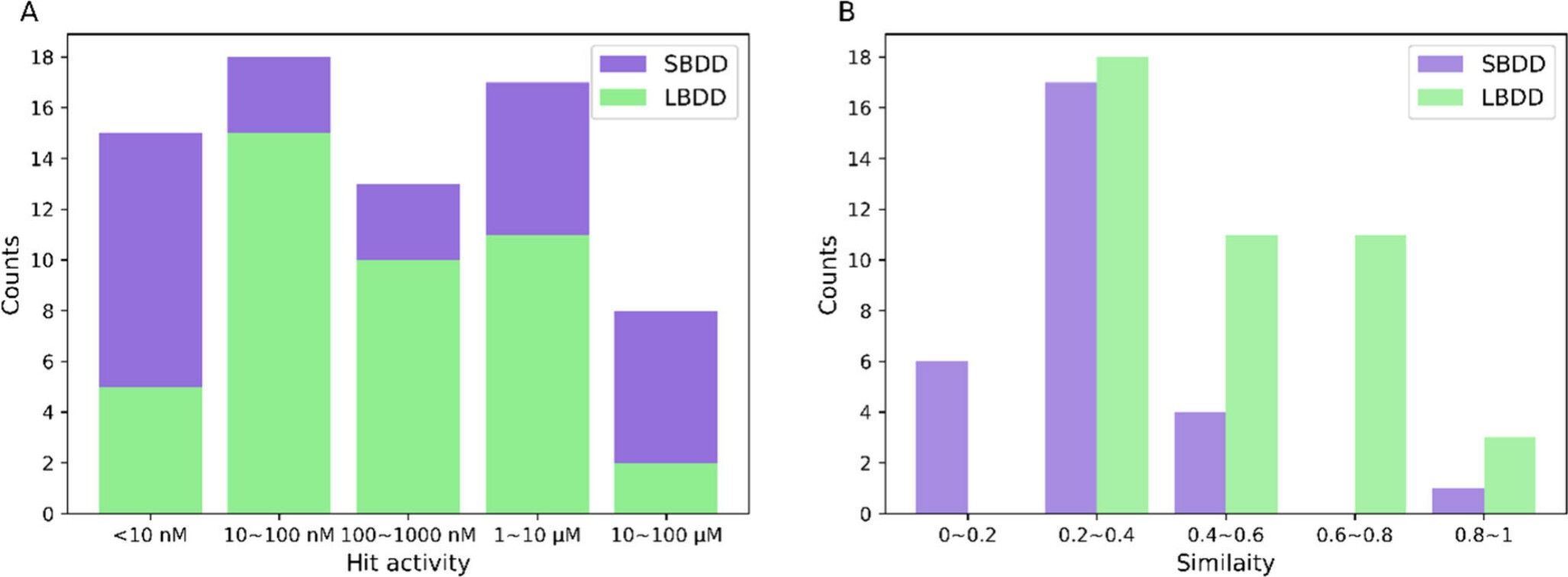
AI设计苗头化合物的效力和结构新颖性分析:(A)所有研究中最佳苗头化合物效力的分布;(B)表示所选苗头化合物结构新颖性的平均Tc(max)值,比较SB与LB方法。图片引自文献10。
解析分子”似曾相识”的根本原因
专家所称的这种”结构同质化”倾向并非单一失误,而是多重因素交织形成的完美风暴的结果。要真正利用这些工具,我们必须首先剖析它们为什么经常将我们引向熟悉的道路。
训练数据的继承偏见:生成式模型首先是其教育的产物。大多数模型都是在像ChEMBL这样的大型公共数据库上训练的,这些数据库不是化学现实的中立、客观集合。相反,它们是反映数十年人类研究优先事项、成功以及最重要的偏见的历史档案。这些数据集严重偏向于”成功的”靶点家族,如激酶和GPCR,并且进一步偏向于通过早期筛选的分子,即具有所谓”类药”性质的分子。模型勤奋地学习这些教训,得出结论认为某些骨架本质上是”好的”,而化学空间的某些区域是不肥沃的,仅仅因为训练数据中没有来自那里的例子。这创造了一个强大的自我强化循环;模型继承了我们的历史焦点,在其输出中过度代表已知骨架,并积极避免真正的探索。它成为已知化学宇宙的大师,生成”Me-too”化合物的专家,但当被要求成为”First-in-class”探索者时,仍然是一个胆小的学徒。
我们衡量标准的局限性:多年来,使用分子指纹(如ECFP4)计算的Tanimoto系数一直是评估化学相似性的主力指标。虽然有用,但过度依赖它进行新颖性评估是一个关键缺陷。这种方法通过将分子分解为一系列小的、局部结构特征来工作。结果是它对表面变化高度敏感。生成式模型可以轻易利用这一弱点:它可以采用一个已知的、已获得专利的骨架,策略性地添加或修改一些外围化学基团(化学家称之为”修饰”),并产生一个低Tanimoto分数的分子,将其标记为”新颖”。然而,在药物化学家训练有素的眼睛看来,它明显是衍生的,完全属于与其母体相同的知识产权空间。这个度量标准从根本上只见树木不见森林,未能识别核心骨架的保守性,而这正是分子身份和IP价值的核心。仅依赖它会产生一种危险的虚假安全感,可能会在死胡同资产上浪费数百万研发资金。
方法论约束和内在权衡:AI模型的架构本身可能会施加其自身的创造性限制。例如,变分自编码器(VAEs)针对编码和解码信息进行了优化,这个过程本质上迫使它们保持接近它们被训练的数据分布,限制了它们真正”分布外”生成的能力。生成对抗网络(GANs)可能更具冒险性,但通常以化学合理性为代价,产生在实验室中不可能创造的合成噩梦。此外,新颖性和可合成性之间存在一个基本的、有充分记录的紧张关系。通常,数学上最新颖的分子也是最奇怪和结构最复杂的,具有张力的环或不稳定的官能团,这会让任何工艺化学家不寒而栗。当我们应用过滤器以确保分子”可合成”时,我们不可避免地将模型推回更熟悉、行为良好的化学模式。这迫使AI在没有明确指导的情况下难以管理的艰难权衡,通常默认选择已知化学的安全性,而不是真正创新的风险。
知识产权死胡同和科学停滞
在学术环境中,生成一个低新颖性的分子可能只是论文中的一个有趣脚注。在工业研发环境中,每个决策都与时间表、预算和市场策略相关联,这种缺乏真正创新的情况会产生严重且连锁的后果。
知识产权风险:任何制药公司的基础资产是其知识产权。发现项目的主要目标不仅仅是找到活性分子,还要确保获得具有清晰且可防御专利的新化学实体(NCE)。浪费数月宝贵的合成和测试资源在一个竞争对手的专利律师可以轻易拆解为”显而易见”或”缺乏创造性步骤”的分子上,相对于现有技术,这是一个灾难性的失败。它将一个有前途的项目变成一个财务黑洞。认识到这一生存威胁后,最先进的团队现在正将自动化专利数据库检查和骨架跃迁指标直接整合到生成工作流程中,知识产权审查不再被视为下游的法律障碍,而是从一开始就作为关键的计算机模拟筛选环节。
资源浪费:DMTA循环的每一个周期都代表着资本和人类专业知识的重要投资——化学家的时间、昂贵的试剂和筛选平台的访问。追求那些仅仅是重新发现已知生物学的化合物,相当于资助一次伟大的探险,却发现它只是绘制了自己总部的地图。更糟糕的是,当AI缺乏历史背景时,无意中复活了公司自己档案中的失败候选物——多年前因毒性、代谢不稳定性或其他看不见的责任而被搁置的化合物。这种由天真的AI应用所导致的”公司健忘症”不仅消耗预算;更严重打击士气的,侵蚀了计算团队和负责合成这些注定失败想法的实验台化学家之间的信任。
错失机会:这可能是最悲惨的后果。AI在药物发现中的最大承诺是它能够找到人类可能错过的优雅、非显而易见的解决方案。我们投资这些技术是为了找到全新的骨架,这些骨架可以解锁新的作用机制,绕过顽固的耐药途径,或实现密切相关的蛋白质家族成员之间的前所未有的选择性。结构同质化直接损害了这一核心使命。这是一种战略失败,使我们固守于现有的化学范式,在我们迫切需要突破时却鼓励渐进主义。它引导我们开发略胜一筹的”同类最佳(Best-in-class)”药物,而真正具有变革性价值的,却在于发现重新定义患者治疗的”同类首创(First—in-class)”药物。
前进的务实路径——要求有价值的新颖性
那么,我们该如何摆脱这种分子“似曾相识”的循环,从生成统计意义上“新颖”的分子转向发现真正有价值的新药呢?这需要一种更复杂、更具怀疑精神和综合性的方法——不是将人工智能视为一个黑箱预言家,而是将其视为一种需要专家操作的强大专业工具。
靶点和方法选择至关重要:战略必须先行于生成!AI方法的选择应由具体的科学问题和项目的战略目标决定。a) 对于一个充分理解的靶点,如果是在已知骨架上进行先导化合物优化,基于配体的方法可能完全足够,甚至更可取。此时的目标不是彻底创新,而是微调,探索R基团修饰或连接臂修饰以增强活性或改善ADME性质。在这种情况下,模型倾向于模仿已知模式是一种特性,而非缺陷。然而,b) 对于几乎没有已知结合剂的新靶点,或者当项目明确需要”骨架跃迁”以逃避竞争对手的知识产权时,基于结构的生成模型变得至关重要。这种方法迫使算法从第一性原理(结合位点的3D几何结构)出发解决问题,大大增加了发现真正新颖且非显而易见的化学骨架的几率。对于这类任务,具有3D感知和/或口袋约束条件的工具是首选。
升级你的新颖性评估工具包:仅依赖单一的Tanimoto打分已不再合理。研发团队必须建立一个稳健的多指标仪表板,全面评估新颖性,并像对待活性或毒性预测一样的严格。这应包括:基于骨架的分析、自动化专利和数据库检索、化学空间分布分析以及强制性的药物化学家审查。像Fréchet ChemNet Distance(FCD)之类的指标有助于回答一个关键问题:模型是否真正在探索新领域,还是只是在其训练数据附近生成密集的分子簇?这为模型的探索能力提供了量化衡量标准。
优先考虑实验验证:一个分子在被制造和测试之前只是一个假设。新颖性和实用性的最终证明不在于计算的打分值,而在于生物测试。最有影响力的成功案例,如MIT Collins实验室发现的结构独特、通过新机起作用的抗生素,完美地印证了这一点。他们的工作没有止步于生成阶段;而是推进到合成和测试,将计算探索建立在坚实的生物现实基础上。这种实验结果的反馈循环对进展至关重要,尤其是失败的结果!为训练和改善下一代模型提供了宝贵的、高质量的数据,使它们更智能,更适应药物发现的复杂需求。
拥抱人机协作(human-in-the-loop)工作流程:最有效的范式不是人机对抗,而是人机协作。正如我在之前的文章中提到的,我们必须将生成式AI视为化学家手头的创意工具,而非科学家的替代者。最佳的工作流程是真正的伙伴关系:AI充当不知疲倦的头脑风暴者,在相同时间内生成人类团队永远无法构想出的成千上万的结构多样且具有合成可行性的创意;自动化过滤器(理想情况下,最好集成在多指标仪表盘中)充当具有怀疑精神的守门人,通过程序化筛选剔除衍生物、已获专利的化合物、不稳定的结构和不可合成的分子;最终由人类药物化学家担任战略决策者和最终裁决者。
炒作之外还有什么?
生成式AI无疑是药物发现工具库的一个强大补充。然而,我们必须抵制那种认为它是自动创新魔法引擎的诱人叙事。从上一代经过充分研究的工具得到的证据清楚地表明,在使用不当的情况下,它们可能引导我们走上老路。它们可以创造出一个令人信服的新颖性幻觉,但在专家的严格审查和严谨的知识产权分析下,这种幻觉很快就会烟消云散。
然而,这一现实检查并不是对AI潜力本身的最终定论。生成化学领域正在以惊人的速度发展。正如我们在前文(第3篇文章:近期AI分子生成器概述)中所讨论的,新一代更复杂的模型已经崭露头角。这些工具信誓旦旦地承诺从一开始就整合合成感知、联合生成具有逆合成路线的3D结合模式,或采用旨在促进探索的更先进学习架构,有望直接应对新颖性挑战。这些新一代方法是否能决定性地克服其前辈们内在的偏见和局限性,仍然是一个悬而未决的关键问题。对其产出的全面回顾性分析仍有待进行,这些承诺是否转化为可专利的有价值化学物质的实际增长,目前尚无定论。
总的来说,这种动态且快速发展的局面最终强化了我们的中心观点。真正的进步不会来自于被动接受AI的输出,而是来自于健康的职业怀疑态度,以及坚定不疑地将这些工具整合到严格的、以人为核心的科学工作流程中的承诺。通过明智地处理我们的数据,要求一个超越简单Tanimoto打分的多维度新颖性视角,并且永不忽视人类化学专家不可或缺的直觉,我们可以积极引导这些强大的算法。我们的角色是引导它们远离仅仅产生”似曾相识”的分子,朝着最终目标前进:发现真正具有突破性的未来药物。
参考文献
- DrugPatentWatch. “An AI Approach to Generate Novel Pharmaceuticals using Patent Data: Revolutionizing Drug Discovery and Navigating Intellectual Property.” DrugPatentWatch Blog, July 17, 2025. https://www.drugpatentwatch.com/blog/
- Handa, Koichi, Morgan C. Thomas, Michiharu Kageyama, Takeshi Iijima, and Andreas Bender. “On the difficulty of validating molecular generative models realistically: a case study on public and proprietary data.” Journal of Cheminformatics 15, no. 112 (2023). doi: 10.1186/s13321-023-00781-1.
- Jarallah, Somayah J., Fahad A. Almughem, Nada K. Alhumaid, Nojoud AL Fayez, Ibrahim Alradwan, Khulud A. Alsulami, Essam A. Tawfik, and Abdullah A. Alshehri. “Artificial intelligence revolution in drug discovery: A paradigm shift in pharmaceutical innovation.” International Journal of Pharmaceutics 680 (2025): 125789. doi: 10.1016/j.ijpharm.2025.125789.
- Jin, Suhyeon, Boram Lee, Kyoungyeul Lee, and Dongsup Kim. “Novelty and Coverage: An Integrated Metric Addressing Quality Trade-offs in AI-Generated Compounds for Drug Discovery.” ChemRxiv (2024). doi: 10.26434/chemrxiv-2024-1sqhr.
- Polykovskiy, Daniil, Alexander Zhebrak, Benjamin Sanchez-Lengeling, Sergey Golovanov, Oktai Tatanov, Stanislav Belyaev, Rauf Kurbanov, Aleksey Artamonov, Vladimir Aladinskiy, Mark Veselov, Artur Kadurin, Simon Johansson, Hongming Chen, Sergey Nikolenko, Alán Aspuru-Guzik, and Alex Zhavoronkov. “Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models.” Frontiers in Pharmacology 11 (2020): 565644. doi: 10.3389/fphar.2020.565644.
- Preuer, Kristina, Philipp Renz, Thomas Unterthiner, Sepp Hochreiter, and Günter Klambauer. “Fréchet ChemNet Distance: A metric for generative models for molecules in drug discovery.” arXiv preprint arXiv:1803.09518 (2018).
- Safizadeh, Hamid, Scott W. Simpkins, Justin Nelson, Sheena C. Li, Jeff S. Piotrowski, Mami Yoshimura, Yoko Yashiroda, Hiroyuki Hirano, Hiroyuki Osada, Minoru Yoshida, Charles Boone, and Chad L. Myers. “Improving Measures of Chemical Structural Similarity Using Machine Learning on Chemical–Genetic Interactions.” Journal of Chemical Information and Modeling 61, no. 9 (2021). doi: 10.1021/acs.jcim.1c00562.
- Venkatraman, Vishwesh, Jeremiah Gaiser, Daphne Demekas, Amitava Roy, Rui Xiong, and Travis J. Wheeler. “Do molecular fingerprints identify diverse active drugs in large-scale virtual screening? (no).” bioRxiv (2022). doi: 10.1101/2022.09.20.508800.
- Visan, Anita Ioana, and Irina Negut. “Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery.” Life 14, no. 2 (2024): 233. doi: 10.3390/life14020233.
- Xie, Shihan, Hui Zhu, and Niu Huang. “AI-Designed Molecules in Drug Discovery, Structural Novelty Evaluation, and Implications.” Journal of Chemical Information and Modeling (2025). doi: 10.1021/acs.jcim.5c00921.
- Zeng, Xiangxiang, Fei Wang, Yuan Luo, Seung-gu Kang, Jian Tang, Felice C. Lightstone, Evandro F. Fang, Wendy Cornell, Ruth Nussinov, and Feixiong Cheng. “Deep generative molecular design reshapes drug discovery.” Cell Reports Medicine 3, no. 12 (2022): 100794. doi: 10.1016/j.xcrm.2022.100794.


