
摘要:Orb V3 是一种基于神经网络势(Neural Network Potential, NNP)的分子力场模型。该模型基于 OMol25 数据集进行训练,其参考数据来源于 ωB97M-V/DEF2-TZVPD 理论水平下的高精度量子化学计算结果,涵盖广泛的分子构型与化学环境,具备良好的泛化能力。本文演示了 Orb V3 力场在小分子几何优化任务中的应用性能,并提供了完整的计算流程、可复现的代码实现及典型算例,以支持其在实际研究中的部署与验证。进一步地,通过扭转角分析(torsion profile )测试,评估了不同计算策略在能量预测精度与计算效率之间的权衡。结果表明,采用 Orb V3 进行几何优化、随后在高精度 DFT 水平(BP86-D3BJ/DEF2-TZVP)进行单点能量校正的“DFT//Orb-V3”策略,能够在显著降低整体计算成本的同时,实现与全 DFT 方法高度一致的能量趋势与相对能垒预测。该方法在保持高精度的前提下展现出优异的计算效率,是一种兼具准确性与实用性的高效计算范式。
简介
Orb V3 是由 Orbital Materials 的 Kareem Abdelmaqsoud 及其同事于 2025 年提出的一种神经网络势(NNP)方法,该方法基于 OMol25 数据集在 ωB97M-V/DEF2-TZVPD 理论水平计算的数据进行训练1,2,3。与专为无机晶体设计的 Orb 1不同,Orb V32 是专为分子体系构建的模型,能够拓展应用于生物学体系,精确模拟金属配合物、生物分子和电解质。此外,Orb V3 还支持对总电荷和自旋多重度进行条件设定,这一关键特性对于许多新型材料与分子的设计具有重要意义。
软件安装
首先安装Orb V3:
1 2 3 4 5 6 7 8 9 10 11 12 | # 创建虚拟环境 cd /public/gkxiao/software python3 -m venv my-orbmol #激活虚拟环境 source my-orbmol/bin/activate #安装Orb V3 pip install orb-models -i https://pypi.tuna.tsinghua.edu.cn/simple #安装cuda支持 pip install --extra-index-url=https://pypi.nvidia.com "cuml-cu12==25.2.*" |
算例
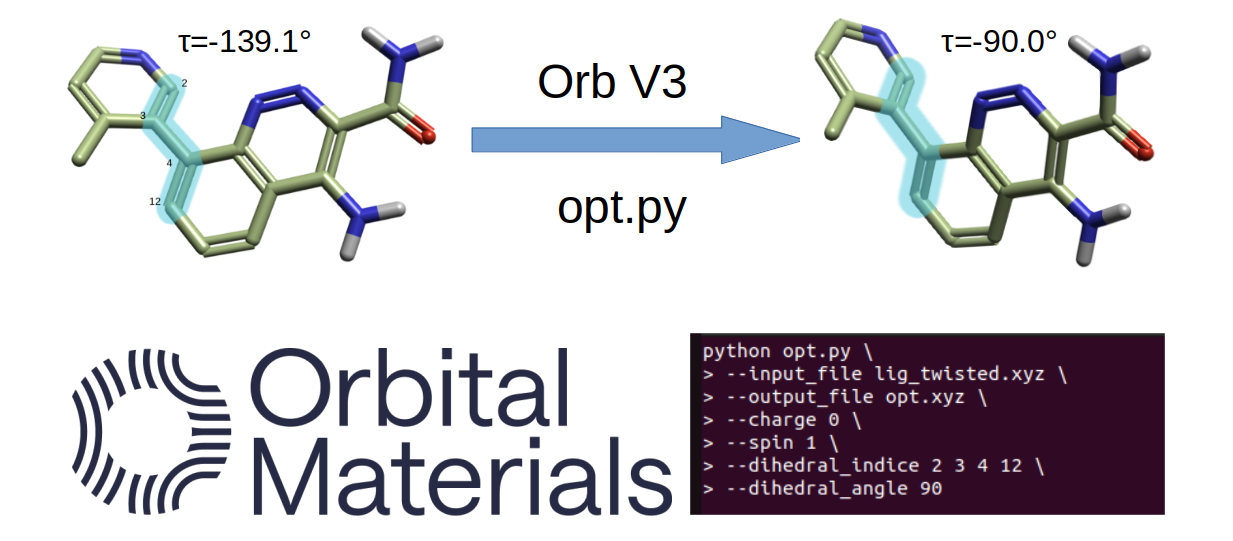
图1. 算例分子是BTK共晶结构PDB 4ZLZ的配体。将其两面角2-3-4-12从生物活性构象-73.8°旋转为-139.1°。
算例分子是BTK共晶结构PDB 4ZLZ的配体,如图1所示。将其两面角2-3-4-12从生物活性构象-73.8°旋转为-139.1°,作为起始构象(lig_twisted.xyz),可从github仓库4获得:
- lig_twisted.xyz:两面角2-3-4-12 = -139.1°
- lig_xtal.xyz:两面角2-3-4-12 = -73.8°
常规几何优化
常规几何优化不对两面角约束:
1 2 3 4 5 | python opt.py --input_file \ lig_twisted.xyz \ --output_file opt.xyz \ --charge 0 \ --spin 1 |
冻结两面角进行几何优化
使用原子索引号(原子序号从1开始)定义两面角,冻结该两面角进行几何优化:
1 2 3 4 5 | python opt.py --input_file opt.xyz \ --output_file opt_frozen.xyz \ --charge 0 \ --spin 1 \ --dihedral_indices 2 3 4 12 |
将两面角约束为指定角度进行几何优化
如果起始结构指定的两面角角度不是你需要的,而是冻结在指定的角度进行几何优化,比如-90°:
1 2 3 4 5 6 | python opt.py --input_file lig_twisted.xyz \ --output_file opt_-90.xyz \ --charge 0 \ --spin 1 \ --dihedral_indices 2 3 4 12 \ --dihedral_angle -90 |
注意:目前脚本只支持对1个两面角进行约束,如果需要对多个两面角约束,需要将这些两面角写成一个list,你需要修改代码。
测试Orb V3:用于扭转角分析
为评估 Orb V3 方法在几何优化与能量计算方面的性能表现,本文开展了一项扭转角扫描分析(torsion profile)计算。其中一个考察的分子结构(Model-1)如图2右上角所示,其扭转二面角由四个高亮原子定义。
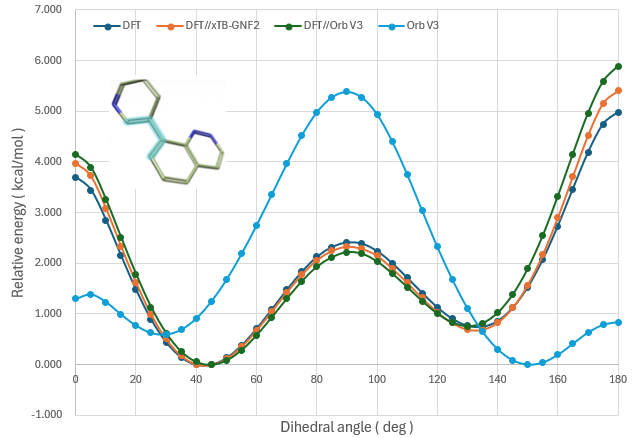
图2. 模型分子Model-1的不同扭转角分析方法的结果比较
在四种不同的理论级别上进行了几何优化与单点能计算:
- Orb V3: 在 Orb V3 理论水平上完成几何优化,并在同一级别进行能量计算;
- DFT//Orb V3:采用 Orb V3 进行几何优化,随后在 BP86-D3BJ/DEF2-TZVP 理论水平上执行单点能量计算;
- DFT//xTB-GFN2:采用 xTB-GFN2 进行几何优化,随后在 BP86-D3BJ/DEF2-TZVP 理论水平上执行单点能量计算;
- DFT:在 BP86-D3BJ/DEF2-TZVP 理论水平上同时完成几何优化与能量计算。
如图2所示,采用 DFT//xTB-GFN2(橘色曲线)以及DFT//Orb-V3 方法(绿色曲线)所获得的扭转角分析曲线与完全基于 DFT 水平(深蓝色曲线)的结果高度一致,三者在整体趋势及相对能量差值方面几乎完全重合。但DFT//xTB-GFN2曲线比DFT//Orb V3曲线又更加靠近完全基于DFT水平的曲线。以DFT方法为参比,计算DFT//Orb-V3与DFT//xTB-GFN2两个方法与参比方法的在各个两面角度上相对能量的差值(取绝对值),进行Wilcoxon检验,结果表明两者在接近完全基于DFT水平方法的程度上有显著差异,W=8.0、p=3.290e-07。以完全DFT方法相比,DFT//xTB-GFN2方法的MAE=0.1156,而DFT//Orb-V3方法的MAE=0.2764,因此DFT//xTB-GFN2方法比DFT//Orb V3方法更接近完全基于DFT的方法。相比之下,仅使用 Orb V3 进行全流程计算(浅蓝色曲线)所得的扭转角分析曲线则表现出显著偏差,尤其在能垒位置和局部极小值分布上存在明显差异。
上述结果表明,在该体系中,Orb V3 能够提供与高精度 DFT 方法相当的几何结构,具备良好的构型描述能力;然而,其能量预测能力与传统密度泛函理论(DFT)存在显著偏差。因此,若仅依赖 Orb V3 进行能量计算,其结果的可靠性值得谨慎评估。值得注意的是,将 Orb V3 用于几何优化、而以高精度 DFT 方法进行后续能量校正(即 DFT//Orb-V3 模型),可在大幅降低计算成本的同时,有效保持能量精度,是一种兼具效率与准确性的可靠计算策略。
代码与数据获取
代码与数据可从github仓库4获得:https://github.com/gkxiao/orb-script
文献
- Mark Neumann, James Gin, Benjamin Rhodes, Steven Bennett, Zhiyi Li, Hitarth Choubisa, Arthur Hussey, Jonathan Godwin. Orb: A Fast, Scalable Neural Network Potential. 2024. arXiv:2410.22570. https://arxiv.org/abs/2410.22570
- Benjamin Rhodes, Sander Vandenhaute, Vaidotas Šimkus, James Gin, Jonathan Godwin, Tim Duignan, Mark Neumann. Orb-v3: atomistic simulation at scale. 2025. arXiv:2504.06231. https://arxiv.org/abs/2504.06231
- https://huggingface.co/Orbital-Materials/OrbMol
- Opt.py. Optimize molecular geometry with optional dihedral angle constraint using ORB v3 force field. Available at: https://github.com/gkxiao/orb-script
- Smith, C. R.; Dougan, D. R.; Komandla, M.; Kanouni, T.; Knight, B.; Lawson, J. D.; Sabat, M.; Taylor, E. R.; Vu, P.; Wyrick, C. Fragment-Based Discovery of a Small Molecule Inhibitor of Bruton’S Tyrosine Kinase. J. Med. Chem. 2015, 58 (14), 5437–5444.


