
基于 Orb V3 神经网络势的几何优化与扭转角分析:方法评估与应用拓展
摘要:本文系统评估了由 Orbmol公司提出的新型神经网络势(Neural Network Potential, NNP)——Orb V3 在分子体系几何优化与构象能计算中的性能表现。通过对典型芳香轴类分子(Model-1 与 Model-2)开展多层级扭转角扫描分析,并对比 DFT//Orb V3、DFT//GFN2-xTB 及全 DFT 方法,本文揭示了 Orb V3 在结构优化能力上的优越性及其在能量预测方面的局限。研究结果表明:虽然 Orb V3 能够提供与高精度 DFT 相当的几何结构描述能力,但其独立执行能量计算时存在显著偏差;而采用“Orb V3 几何优化 + 高精度 DFT 单点能校正”的混合策略(DFT//Orb V3)可有效实现效率与准确性的平衡。此外,通过对 BTK 抑制剂系列化合物的构象分析,我们进一步验证了芳香轴邻位甲基取代所引发的构象锁定效应,并将其归因于立体应变能的降低,从而为理解分子结构—活性关系提供了理论依据。
肖高铿/2025-09-28
1. Orb V3 神经网络势概述
Orb-v3 是 Orbital Materials 公司推出的新一代通用机器学习原子间势函数(MLIP),在性能-速度-内存的帕累托前沿上实现显著突破1-3。该模型系列包括多个变体,涵盖从高精度保守型到高效率稀疏图非保守型的不同配置,适用于广泛的计算化学任务。本文将重点考察Orb V3在构象能面构建中的应用潜力,并与传统方案(如 DFT//GFN2-xTB)进行系统比较。
2. 方法与模型设置
2.1 模型分子设计
为评估 Orb V3 的性能,我们选取两类代表性芳香轴结构作为研究对象:
- Model-1:原始无取代芳香轴分子,用于考察基础构象自由度。
- Model-2:在Model-1 吡啶环邻位引入甲基取代基的衍生物,模拟实际药物分子中常见的立体阻碍效应。
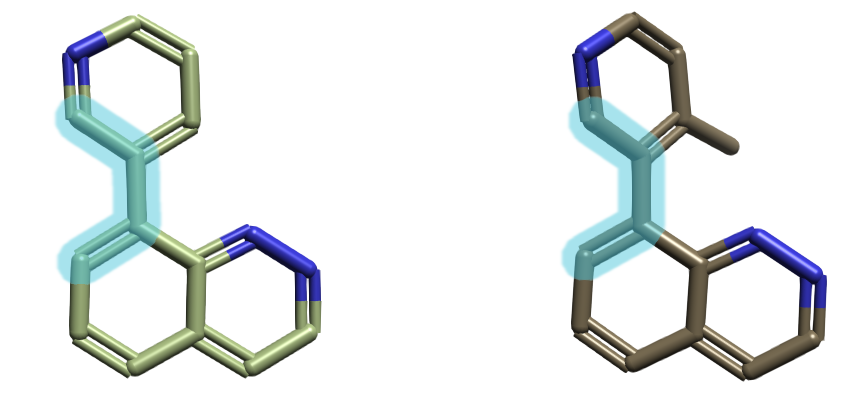
图1. 模型分子Model-1(左)与Model-2(右)的化学结构式。
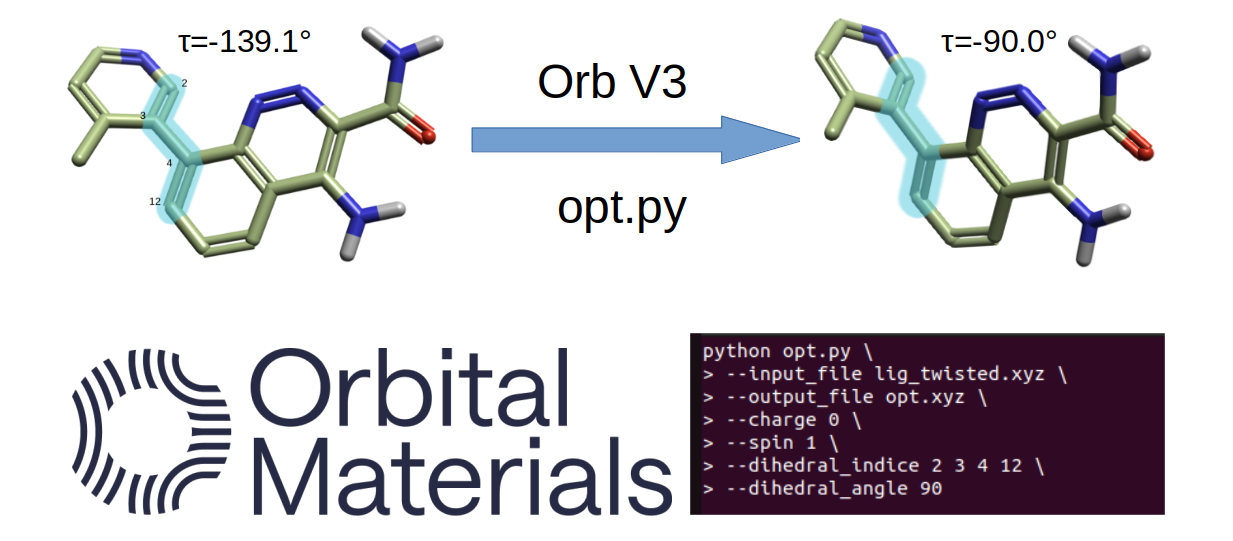
两个分子均以连接两个芳香环的单键作为旋转坐标(Ar–Ar 轴),进行 0° 至 180° 的扭转角扫描,步长为 5°,每构象点通过几何优化后执行能量计算。
2.2 多层级理论方法对比
在四种不同的理论级别上进行了几何优化与单点能计算:
- Orb V3: 在 Orb V3 理论水平上完成几何优化,并在同一级别进行能量计算;
- DFT//Orb V3:在 Orb V3 理论水平进行几何优化,随后在 BP86-D3BJ/DEF2-TZVP 理论水平上执行单点能量计算;
- DFT//GFN2-xTB:在 GFN2-xTB 理论水平进行几何优化,随后在 BP86-D3BJ/DEF2-TZVP 理论水平上执行单点能量计算;
- DFT:在 BP86-D3BJ/DEF2-TZVP 理论水平上同时完成几何优化与能量计算。
所有的DFT计算均在 ORCA (Version 6.01) 环境中执行,GNF2-xTB在xTB (Version 6.7.1)环境中执行,Orb V3通过自定义python脚本opt.py4处理。
3. 构象能分析与方法评估
3.1 Model-1 的扭转势能面
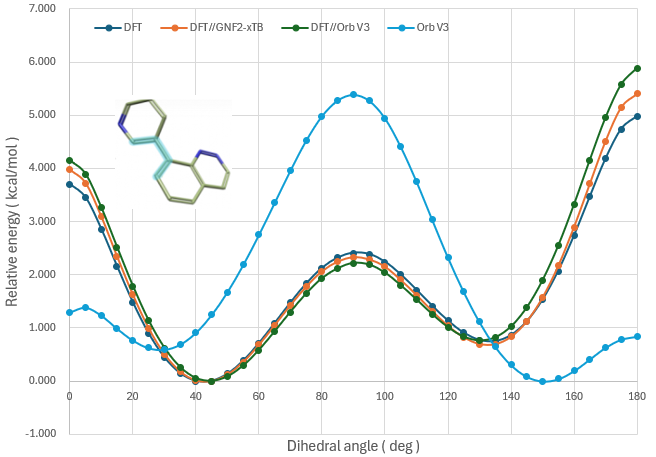
图2. 模型分子Model-1的不同扭转角分析方法的结果比较
在DFT(BP86-D3BJ/DEF2-TZVP)理论水平下对Model-1进行的扭转角扫描分析结果(图2)表明,其分子内旋转势能曲线整体变化相对平缓。全局能量最小值出现在二面角约为40°的构象(能量为0.000 kcal/mol),同时在130°附近存在一个局部极小点(能量为0.769 kcal/mol),二者均为较稳定的低能构象。在两个极小值之间,约90°处存在一过渡态,其能垒高度为2.41 kcal/mol,这一数值较低,表明在室温条件下分子可克服该能垒,实现构象间的快速互变。此外,Model-1在二面角为0°与180°时能量最高,分别达到3.70 kcal/mol与4.98 kcal/mol(约5.0 kcal/mol),这可视为吡啶单元绕芳香轴旋转所需克服的最高能垒。该能垒远低于一般认为的21 kcal/mol的构象限制阈值(一般认为高于此值则旋转受阻),进一步说明该分子具有较高的构象柔性,其在室温下可发生快速的绕轴旋转运动。
表1. 两种混合扭转角分析方法与全DFT基准方法在构象能计算中的偏差统计(单位:kcal/mol)
| 误差类型 | DFT//GFN2-xTB | DFT//Orb V3 |
|---|---|---|
| MAE | 0.116 | 0.276 |
| MedE | 0.084 | 0.193 |
MAE: 平均绝对误差; MedE:中位数误差(Median Error)
图2的扭转角分析结果显示,DFT//GFN2-xTB(橙色曲线)与DFT//Orb-V3(绿色曲线)的计算结果与基准DFT方法(深蓝色曲线)高度吻合,不仅在整体能量变化趋势上一致,在相对能量差值方面也几乎完全重合。值得注意的是,DFT//GFN2-xTB曲线相较于DFT//Orb-V3曲线更接近DFT参考基准。以DFT方法为基准,Wilcoxon符号秩检验表明:DFT//Orb-V3和DFT//GFN2-xTB在绝对误差与基准方法的接近程度上存在统计学显著差异(W = 8.0,p = 3.290 × 10⁻⁷)。其中,DFT//GFN2-xTB的平均绝对误差(MAE = 0.116)显著低于DFT//Orb-V3(MAE = 0.276),详细数据见表1。相比之下,仅使用Orb-V3完成全流程计算(图2浅蓝色曲线)所得结果与DFT基准存在显著偏差,尤其在能垒高度和局部极小值分布方面表现出明显差异。
3.2 Model-2 的扭转势能面:探索立体效应对构象的影响
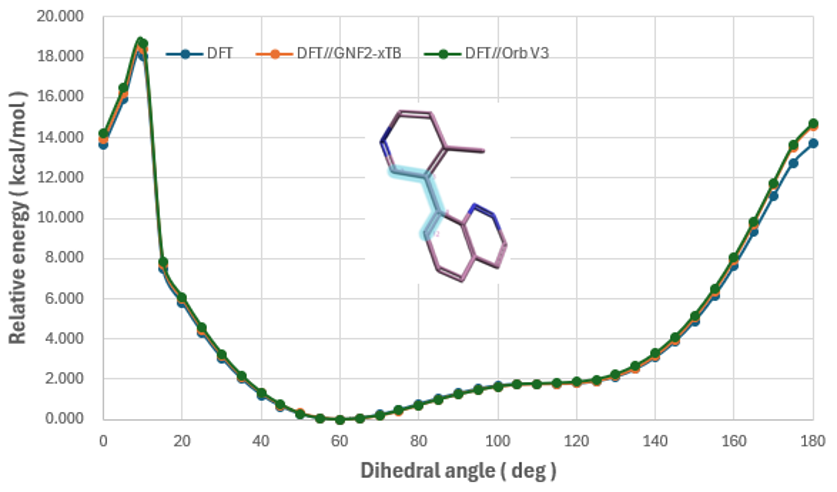
图3. 模型分子Model-2的扭转角分析结果
引入邻位甲基后,Model-2 的扭转角分析结果(图3)表现出显著的构象锁定效应:
- 在 0° 和 180° 处分别存在高达 13.698 与 18.081 kcal/mol 的能量壁垒;
- 唯一稳定极小值出现在 60° 构象,在+/-15° 范围内,能量低于 0.65 kcal/mol。
该结果清晰展现了甲基引入所造成的空间排斥,导致芳香轴旋转受到严重限制。
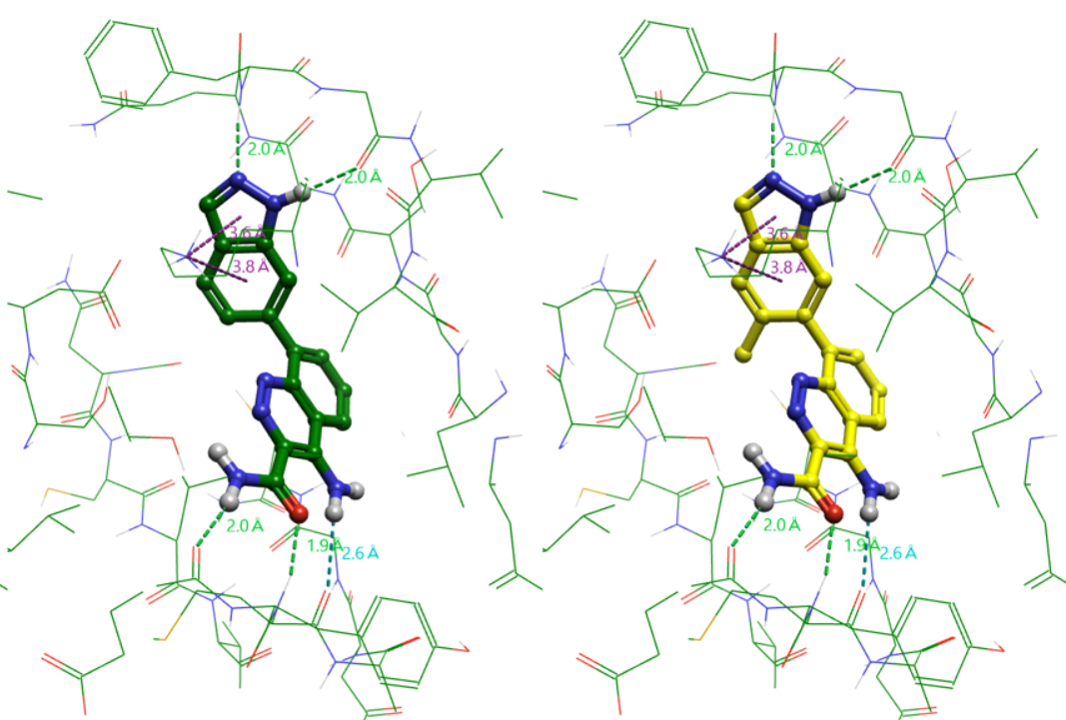
图4. Smith 等人5报道的 BTK 抑制剂化合物 10(左)与 11(右)在 PDB 4ZLZ 活性位点中的结合模式(Flare 分子对接结果)
这一构象锁定效应在 Smith 等人5报道的 BTK 抑制剂体系中得到验证(图4):
- 化合物 10 与 11 结构高度相似,仅苯环邻位是否存在一个甲基取代基;
- 晶体结构 PDB 4ZLZ 显示二者在活性口袋内结合模式一致,均呈现 –73.8° 的芳香轴二面角;
- 然而其生物活性存在显著差异:化合物 10 IC50 = 12 nM,化合物 11 IC50 = 4 nM(活性提升约 3 倍)。
根据前述对模型分子 Model-1 与 Model-2 在 DFT(BP86-D3BJ/DEF2-TZVP)理论水平下的扭转角扫描结果(图3),推测结合构象张力能分别为:
- 化合物 10:1.83 kcal/mol;
- 化合物 11:0.47 kcal/mol。
该能量差异部分解释了其生物活性差异:邻位甲基诱导构象锁定,有效降低了结合态下的构象张力能,从而增强与靶点的亲和力。
进一步地,基于 DFT//Orb V3 与 DFT//GFN2-xTB 的估算结果与全 DFT 趋势高度一致(图3与表2),验证了该混合策略在量化构象应变能方面的可靠性。而独立的Orb V3计算的相对能量值严重偏离基准方法(参见数据部分的扭转角分析结果文件)。
表2. 两种混合扭转角分析方法与全DFT基准方法在构象能计算中的偏差统计(单位:kcal/mol)
| 误差类型 | DFT//GFN2-xTB | DFT//Orb V3 |
|---|---|---|
| MAE | 0.163 | 0.237 |
| MedE | 0.071 | 0.135 |
MAE: 平均绝对误差; MedE:中位数误差(Median Error)
4. 讨论:Orb V3 的适用场景与局限性
尽管 Orb V3 在几何优化方面表现出色,能够精确捕捉复杂分子的稳定构象及局部势能特征,但在独立进行能量预测时仍存在系统偏差。尤其在涉及高精度相对能差(如构象稳定性、结合亲和力差异)计算中,应避免直接使用 Orb V3 单点能。
相比之下:
- DFT//GFN2-xTB被广泛验证为“速度与精度的最佳平衡”方案5,在自由能计算(FEP)、分子动力学(MD)模拟及新分子自定义力场参数化中表现出极强的适用性6;
- DFT//Orb V3亦具备良好前景,尤其适合在大规模构象空间探索中作为初步筛选工具。
综合来看,推荐使用“Orb V3 几何优化 + DFT 单点校正”或“GFN2-xTB 几何优化 + DFT 能量校准”的工作流,在保证效率的同时维持足够精确度。
5. 总结
本研究系统评估了 Orb V3 力场在分子构象分析中的表现:
- Orb V3 在几何结构优化方面表现出色,可准确复现 DFT 水平下的稳定构象;
- 其独立能量预测存在偏差,不建议直接用于精确相对能计算,尤其在torsion profiling这类的势能面分析场景需要小心谨慎;
- 与 GFN2-xTB 相比,DFT//Orb V3 能力相近,但在某些非键相互作用描述上更具优势;
- 通过模型分子与真实药物体系的关联分析,证实了邻位甲基诱导的构象锁定效应可通过降低张力能提升结合亲和力,为合理药物设计提供理论依据。
6. 数据与代码获取
本研究相关脚本公开在 GitHub 仓库中,欢迎下载复现:https://github.com/gkxiao/orb-script
项目包含以下内容:
文献
- Mark Neumann, James Gin, Benjamin Rhodes, Steven Bennett, Zhiyi Li, Hitarth Choubisa, Arthur Hussey, Jonathan Godwin. Orb: A Fast, Scalable Neural Network Potential. 2024. arXiv:2410.22570. https://arxiv.org/abs/2410.22570
- Benjamin Rhodes, Sander Vandenhaute, Vaidotas Šimkus, James Gin, Jonathan Godwin, Tim Duignan, Mark Neumann. Orb-v3: atomistic simulation at scale. 2025. arXiv:2504.06231. https://arxiv.org/abs/2504.06231
- https://huggingface.co/Orbital-Materials/OrbMol
- Opt.py. Optimize molecular geometry with optional dihedral angle constraint using ORB v3 force field. Available at: https://github.com/gkxiao/orb-script
- Smith, C.R. et al. (2015) “Fragment-Based Discovery of a Small Molecule Inhibitor of Bruton’s Tyrosine Kinase,” Journal of Medicinal Chemistry, 58(14), pp. 5437–5444. Available at: https://doi.org/10.1021/acs.jmedchem.5b00734.
- 使用半经验GFN2-xTB计算来处理含100多个原子较大分子结构的能量最小化. http://blog.molcalx.com.cn/2023/08/05/minimization-using-semi-empirical-gfn2-xtb.html
- 用DFT//GFN2-xTB 混合方法自定义力场参数. http://blog.molcalx.com.cn/2023/08/06/customized-force-field-parameters-dftgfn2-xtb.html


