
查寻分子的准备对基于形状虚拟筛选的影响
摘要:基于形状的虚拟筛选已成为从超大型化合物库中识别结构新颖生物活性分子的有力策略。然而,此类方法的性能对查询分子的构象状态高度敏感。本研究以阿斯利康最新报道的ROS1选择性抑制剂化合物31为模型系统,探讨查询构象准备对虚拟筛选结果的影响。通过量子力学优化以及与多个ROS1共晶配体(PDB: 3ZBF, 4UXL, 7Z5X, 7Z5W)的结构比对,我们鉴定出一个能重现蛋白质-配体复合物中关键铰链区和DFG区相互作用的低能生物活性构象(CONF_34)。与之相对,我们选取了一个高能构象(CONF_17,ΔE = 8.5 kcal/mol)代表能量不可及状态的非生物活性构象。使用ROSHAMBO2平台并采用Tanimoto和Tversky相似性度量,将这两个构象作为查询对源自ChEMBL35的精选3D构象集合(约135万化合物,约2780万构象体)进行基于形状的虚拟筛选。尽管全局得分分布相近,但生物活性构象在检索已知ROS1抑制剂方面显著优于非生物活性构象:在标准竞争排序下,高亲和力先导化合物CHEMBL1997924(Ki = 1 nM)使用CONF_34时排名第11位(RefTverskyCombo = 1.88),而使用CONF_17时排名第50位(RefTverskyCombo = 1.81)。此外,基于Tversky的评分(α = 0.95)展现出比传统Tanimoto组合评分更优异的排序性能。这些结果强调,查询构象选择不仅是预处理步骤,更是虚拟筛选效能的决定性因素。我们主张在基于形状的筛选流程中查询分子使用生物活性相关的构象,以提升新型高质量先导化合物的早期发现效率。
1. 前言
阿斯利康(AstraZeneca)的Petrović等人1报道了一项基于OpenEye FastROCS2的虚拟筛选研究。该方法通过在云端部署的数百个GPU并行计算,对阿斯利康虚拟化合物库中规模达1010的分子子集进行了系统性筛选。研究团队进一步对识别出的潜力骨架进行结构枚举,探索了涵盖约1015个分子的完整虚拟化学空间。对FastROCS筛选获得的苗头化合物进行了分子对接分析,并从中选取两个系列进行化学合成及生物学评价,测定其对ROS1与TrkA激酶的抑制活性。该研究展示了基于形状相似性的筛选技术3在超大规模化学库中发现新型、高活性及高选择性抑制剂的可行性与实用价值。
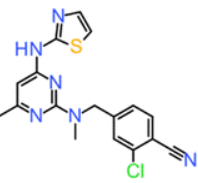
图1. 化合物31的化学结构式
在该研究中,化合物31(结构见图1)被鉴定为一类具有代表性的ROS1选择性抑制剂。该化合物在体外表现出显著的激酶选择性:对ROS1的IC50值为97 nM,而对TrkA的IC50值大于100 μM。值得注意的是,截至本文撰写时,在ChEMBL数据库4中以化合物31为查询结构,采用Morgan指纹(半径=2,指纹长度2048位)进行相似性搜索(Tanimoto相似度阈值\(≥\)0.4),未发现任何结构相似的已知化合物。这一结果进一步凸显了基于形状的虚拟筛选技术在发现结构新颖先导化合物方面的优势。
查询分子(Query)的构建策略对基于形状的虚拟筛选效果具有重要影响。已有大量研究对领先的形状匹配技术ROCS5进行了系统探讨,相关文献我们已在前期工作中予以综述6。本文旨在以化合物31的发现为例,探讨查询构象选择对虚拟筛选结果的影响。需要说明的是,由于原始筛选数据库不可公开获取,且所需计算资源庞大,完全重复Petrović等人1的研究既不现实也无必要。因此,本研究选择化合物31作为查询结构,在ChEMBL数据库中进行虚拟筛选,系统评估查询分子准备对筛选结果的影响,重点考察已知ROS1抑制剂的命中情况及其在不同查询构建策略下的排名变化。
2. 结果与讨论
2.1 查询分子的构象准备:生物活性构象识别
本研究选取Petrović等人2在虚拟筛选中采用的四个ROS1共晶结构(PDB编号:3ZBF、4UXL、7Z5X、7Z5W)中的配体作为参考分子。通过Flare软件中的Conf Hunt & Align模块,将化合物31以”Accurate but slow”模式与上述参考分子进行构象叠合分析。
为确保所选构象具有合理的能量特征,对Conf Hunt & Align模块获得的构象进行了系统的量子力学优化。首先在GFN2-xTB理论水平完成几何优化,随后在R2SCAN-3c//DEF2-mTZVPP理论水平进行单点能计算。以均方根偏差(RMSD)阈值0.125 Å作为构象聚类标准,最终获得包含133个构象的子集(详见数据部分:31_CONFS_QM.sdf)。
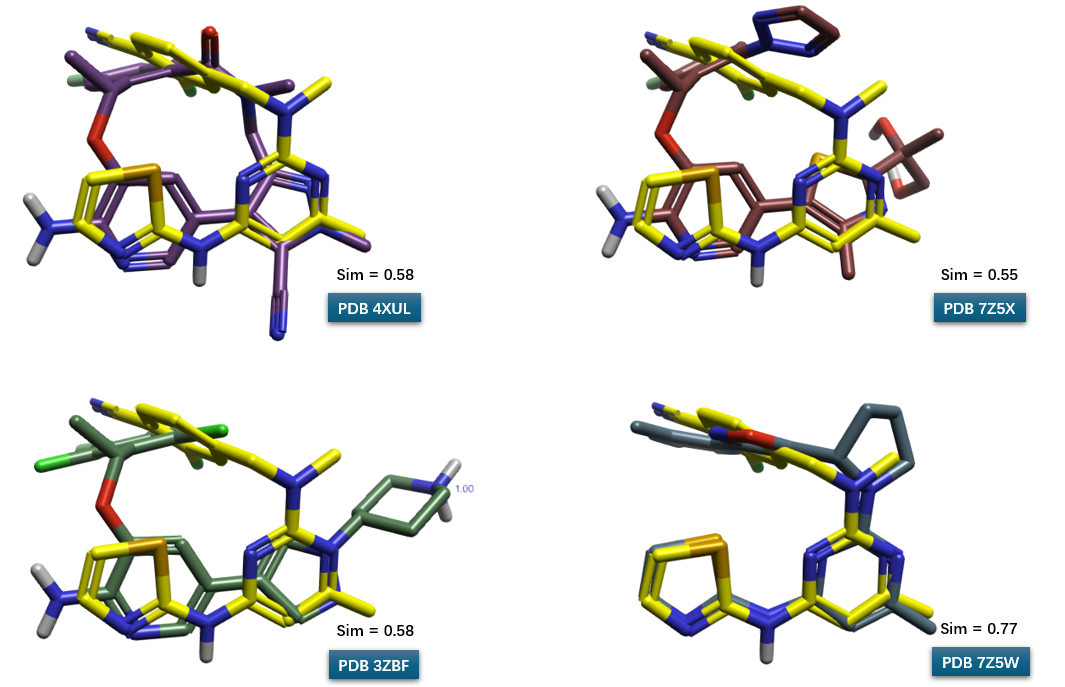
图2. 化合物31与四个ROS1共晶结构(PDB 3ZBF、4UXL、7Z5X、7Z5W)中配体的三维叠合结果。黄色结构代表化合物31,其他颜色分别代表各共晶结构中的参考配体。Smi表示Flare XED场点相似性评分。
鉴于化合物31与PDB 7Z5W共晶配体具有共同的铰链区结合片段,叠合结果显示该公共子结构完全重合(图2右下),且整体叠合相似性评分较高(Sim \(>\) 0.74)。系统分析所有叠合结果发现,共有7个低能构象(构象能范围0.836–1.137 kcal/mol,编号CONF_30、34、45、48、36、41、46)以高度一致的方式与PDB 7Z5W共晶配体叠合。进一步将这7个构象归类为两个代表性构象簇,分别以CONF_34和CONF_41为代表,其主要差异源于3-氯-4-腈基苯环的构象翻转导致的氯原子空间取向不同。图2(右下)展示了化合物31的CONF_34构象。
将化合物31分别与另外三个ROS1共晶结构(PDB 3ZBF、4UXL、7Z5X)的配体进行叠合分析,结果表明CONF_34/41构象能够同时与这三个共晶配体实现良好匹配,其叠合视觉效果如图2所示。
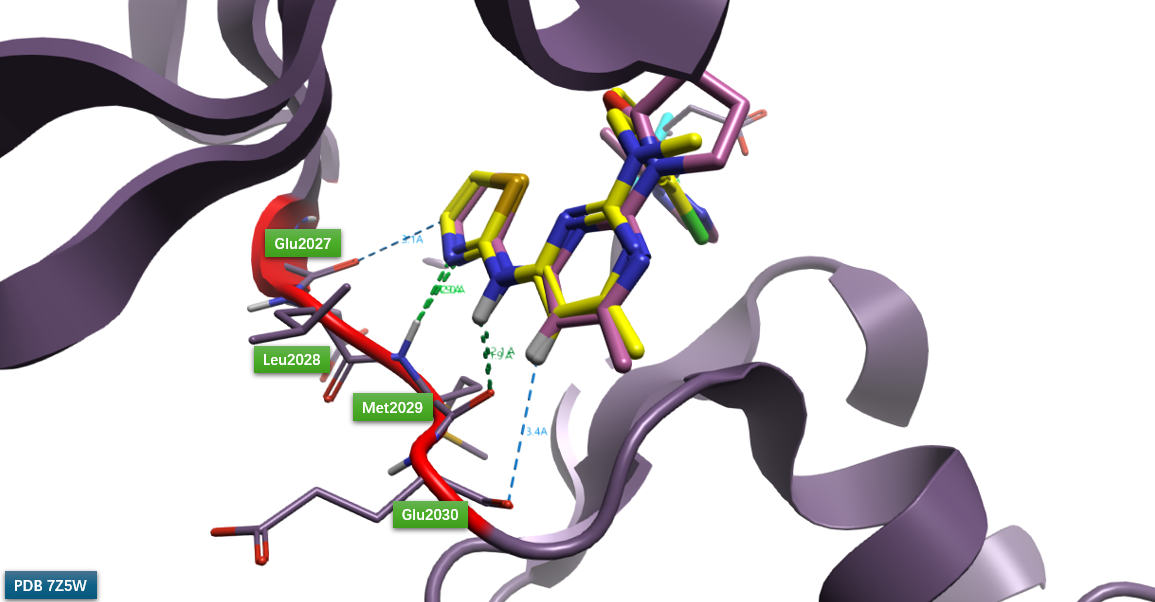
图3. 化合物31与PDB 7Z5W共晶配体在ROS1铰链区的结合模式比较。飘带图表示ROS1蛋白结构,红色区域为激酶铰链区,青色区域为DFG片段;黄色棍棒表示化合物31,紫色棍棒表示PDB 7Z5W共晶配体。
在ROS1结合口袋(基于PDB 7Z5W结构)中观察化合物31的结合模式,如图3所示,化合物31与PDB 7Z5W共晶配体在激酶铰链区(红色飘带)展现出完全一致的结合特征。噻唑环、嘧啶环及其连接臂NH基团构成共平面结构,嘧啶氮与噻唑氮之间的S···N超共轭效应有助于稳定该平面构型。具体而言,噻唑氮作为氢键受体与铰链区Met2029的酰胺NH形成氢键;连接两个芳香环的NH基团作为氢键供体,与Met2029的羰基氧形成氢键。此外,噻唑与嘧啶环上的C–H基团可分别与Glu2027和Glu2030的羰基氧形成C–H···O=C型氢键相互作用。
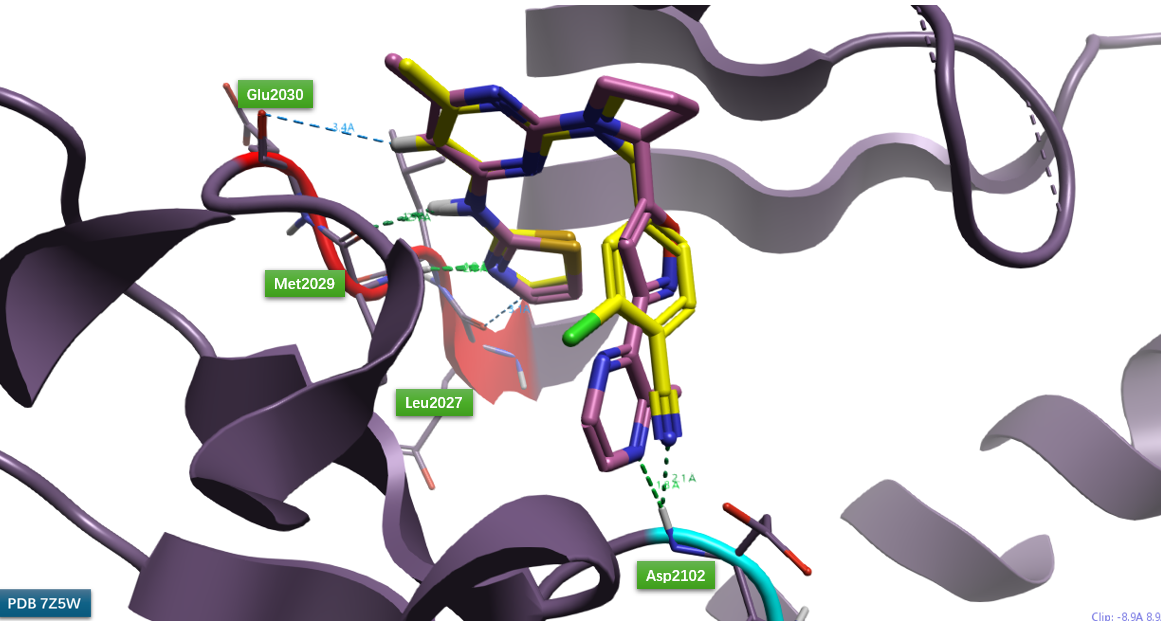
图4. 化合物31与PDB 7Z5W共晶配体在ROS1 DFG区的结合模式比较。飘带图表示ROS1结构,红色为铰链区,青色为DFG片段;黄色棍棒为化合物31,紫色为共晶配体。
如图4所示,化合物31的3-氯-4-腈基苯片段与PDB 7Z5W共晶配体中的异噁唑片段在空间上高度重合。苯环对位的腈基朝向结合口袋中的DFG残基(青色飘带)延伸,其氮原子与共晶配体中哒嗪环上的氮原子位置重合,并作为氢键受体与DFG片段中Asp2012的酰胺NH形成较强的氢键作用。
连接化合物31铰链区结合片段与DFG区结合片段(3-氯-4-腈基苯基)的N,N-二甲氨基结构单元,与PDB 7Z5W共晶配体中的四氢吡咯环在空间上完全重合(图4),可视作对四氢吡咯环的开环模拟。
综上所述,本研究获得的化合物31构象具备生物活性构象的关键特征:1)构象张力能较低;2)与ROS1共晶结构PDB 7Z5W中的配体在形状与静电性质上高度相似(Sim = 0.77);3)在ROS1激酶结合口袋的铰链区与DFG区均能复现共晶配体关键相互作用;4)连接两个功能片段的连接臂结构有效模拟了共晶配体中的对应片段。上述结果表明,该构象可作为可靠的生物活性构象,适用于后续虚拟筛选研究。
2.2 非活性构象的设计策略
为评估非活性构象对虚拟筛选结果的影响,从结构准备过程中排除的高能构象中选取CONF_17作为非活性构象代表,其具备以下特征:1)能量不可及性,构象张力能为8.5 kcal/mol(基于B3LYP-D3BJ/6-31g+(d)//GFN2-xTB理论水平计算);2)与推测的生物活性构象CONF_34存在显著差异,如图5所示:铰链结合片段的噻唑环发生构象翻转;与DFG区相互作用的3-氯-4-腈基苯基发生苯环旋转;连接铰链结合片段与DFG相互作用片段的N,N-二甲基连接臂平面发生构象翻转。
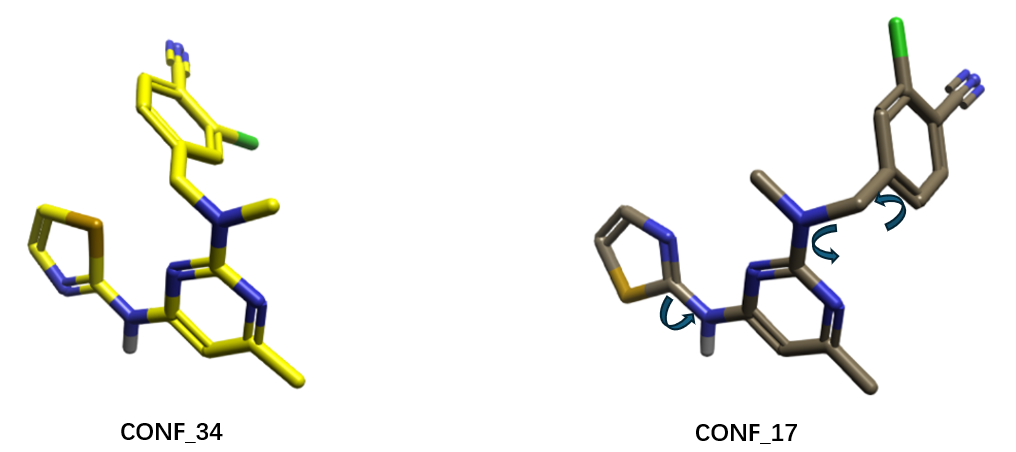
图5. 化合物31推测的活性构象(CONF_34)与非活性构象(CONF_17)的结构比较
2.3 ChEMBL35化合物库的构建
从ChEMBL35数据库7以SMILES格式导出原始数据,经过标准化、脱盐、元素过滤(保留仅含C、H、O、N、S、P、卤素的分子)、去重、类药性过滤及官能团过滤(去除反应性基团)等步骤获得初始化合物库。随后在固定pH 7.4条件下进行质子化状态处理,采用CDPKit的Confgen模块8,9生成三维构象(包括对未明确立体化学中心进行枚举),通过构象分析获得构象系综(每个分子保留能量最低的25个构象)。整个处理过程中化合物数量与构象变化汇总如表1所示。
| 处理阶段 | 数量 |
|---|---|
| 初始化合物 | 910,625 |
| 处理后化合物 | 1,352,233 |
| 生成构象总数 | 27,798,929 |
2.4 虚拟筛选
本研究采用近期公开的ROSHAMBO2方法10,11作为虚拟筛选引擎,该方法基于与ROCS相似的三维形状匹配技术,对ChEMBL35数据库进行系统性筛选。在搜索过程中,数据库中各化合物的构象通过刚性优化算法与查询结构进行三维形状叠合,以实现最大体积重叠。分子间相似性通过形状和药效团特征(即颜色特征)的Tanimoto评分进行量化,各项评分取值范围为0至1。形状与药效团评分之和记为Tanimoto_combo_legacy(Tc)评分,其取值范围为0至2,数值越高表明化合物相似性越强。ROSHAMBO2中引入的形状与颜色归一化组合评分tanimoto_combination,其物理意义与Tc评分一致,但已归一化至0-1范围。值得注意的是,当查询分子与数据库分子存在显著尺寸差异时,偏向查询分子或数据库分子的Tversky评分通常表现出更优的性能12,13。因此,本研究额外引入RefTverskyCombo与FitTverskyCombo评分,分别代表偏向查询分子(α=0.95)与偏向数据库分子的Tversky组合评分。
在本研究设计中,ROSHAMBO2分别采用化合物31的生物活性构象(CONF_34)与非活性构象(CONF_17)对ChEMBL35数据库进行筛选,输出tanimoto_combination(下文简称Tc)评分最高的10,000个化合物。对这些化合物进行聚类分析,对于具有相同名称的化合物,选取其RefTverskyCombo评分最高的异构体作为代表,用以表征其与化合物31的三维结构相似性。基于该相似性值对化合物进行降序排列,构建计算苗头化合物列表。通过检索ChEMBL数据库,系统考察列表中首个具有ROS1抑制活性的化合物在排序中的位置及其评分值,为后续分析提供依据。
| Items | Tanimoto Combo (Legacy) | Tanimoto Combination | RefTversky Combo | FitTversky Combo |
|---|---|---|---|---|
| count | 10000 | 10000 | 10000 | 10000 |
| mean | 1.21 | 0.60 | 1.49 | 1.52 |
| std | 0.05 | 0.03 | 0.11 | 0.12 |
| min | 1.16 | 0.58 | 1.19 | 1.20 |
| 25% | 1.17 | 0.59 | 1.42 | 1.44 |
| 50% | 1.19 | 0.60 | 1.49 | 1.52 |
| 75% | 1.23 | 0.61 | 1.57 | 1.60 |
| max | 1.57 | 0.78 | 1.97 | 2.11 |
化合物31活性构象(CONF_34)与非活性构象(CONF_17)虚拟筛选前10,000个化合物的四个关键组合评分描述性统计分别如表2与表3所示。从评分值的统计分布特征分析,两种构象的筛选结果未表现出显著差异。
| Items | Tanimoto Combo (Legacy) | Tanimoto Combination | RefTversky Combo | FitTversky Combo |
|---|---|---|---|---|
| count | 10000 | 10000 | 10000 | 10000 |
| mean | 1.22 | 0.61 | 1.54 | 1.50 |
| std | 0.04 | 0.02 | 0.10 | 0.11 |
| min | 1.18 | 0.59 | 1.21 | 1.24 |
| 25% | 1.19 | 0.60 | 1.47 | 1.42 |
| 50% | 1.21 | 0.61 | 1.54 | 1.48 |
| 75% | 1.24 | 0.62 | 1.61 | 1.56 |
| max | 1.53 | 0.77 | 1.96 | 2.10 |
在前10,000个高评分化合物中,CONF_17仅识别出1个已知ROS1抑制剂:
- CHEMBL1997924: Tc = 1.33,RefTc = 1.81,ROS1 Ki = 1 nM
而CONF_34成功识别出三个已知ROS1抑制剂:
- CHEMBL1997924: Tc = 1.43,RefTc = 1.88,ROS1 Ki = 1 nM
- CHEMBL1970189: Tc = 1.21,RefTc = 1.68,ROS1 Ki = 19.95 nM
- CHEMBL1983923: Tc = 1.22,RefTc = 1.49,ROS1 Ki = 199.53 nM
这一观察结果具有一定启示性:尽管活性构象与非活性构象均能以相近的评分值召回高活性化合物CHEMBL1997924,但活性构象展现出更优的识别能力。
为深入评估排序性能,本研究采用Pandas的rank()函数实施两种排序策略:
- min(标准竞争排名):并列条目获得相同的最低可能排名,后续排名跳过并列条目所占用的名次。示例:1, 2, 2, 4, 5,…(两个第2名,下一个为第4名)
- dense(密集排名):并列条目获得相同的排名,后续排名保持连续不跳位。示例:1, 2, 2, 3, 4,…(两个第2名,下一个为第3名)
值得注意的是,无论采用活性构象(CONF_34)还是非活性构象(CONF_17)进行虚拟筛选,命中的评分最高的已知ROS1抑制剂均为化合物CHEMBL1997924(Ki = 1 nM)。
| Query | RefTversky Combo | Min Rank | Dense Rank | Index position |
|---|---|---|---|---|
| CONF_34 | 1.88 | 11 | 11 | 51 |
| CONF_17 | 1.81 | 50 | 36 | 298 |
以RefTversky Combo作为评分函数时,采用活性构象(CONF_34)的虚拟筛选结果展现出显著更优的排名表现(表4):在标准竞争排名(min rank)中,活性构象排名第11位,而非活性构象仅排名第50位。
| Query | Tanimoto Combo | Min Rank | Dense Rank | Index position |
|---|---|---|---|---|
| CONF_34 | 1.43 | 52 | 52 | 51 |
| CONF_17 | 1.33 | 299 | 298 | 298 |
同样地,以Tc作为评分函数时,活性构象(CONF_34)的虚拟筛选结果同样表现出显著优势(表5):在标准竞争排名中,活性构象排名第52位,而非活性构象仅排名第299位。此外,研究还发现Tc评分对化合物的排序效果不如RefTversky评分理想,这表明在此特定案例中,Tversky评分相较于传统Tc评分具有明显优势。
2.5 活性构象(CONF_34)虚拟筛选结果的视觉分析(Visual Inspection)
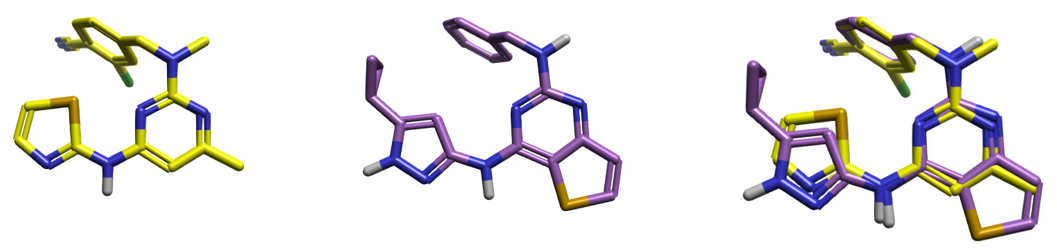
图6. 化合物31活性构象与苗头化合物CHEMBL1997924的构象叠合分析。黄色:化合物31的活性构象CONF_34;紫色:苗头化合物CHEMBL1997924。
视觉分析(Visual Inspection)是虚拟筛选后处理,尤其是决策阶段,不可或缺的关键环节14。如图6所示,化合物13的活性构象与CHEMBL1997924展现出显著的空间匹配特征。从整体构象来看,两个分子的重原子在空间排布上表现出高度一致性(图6右)。
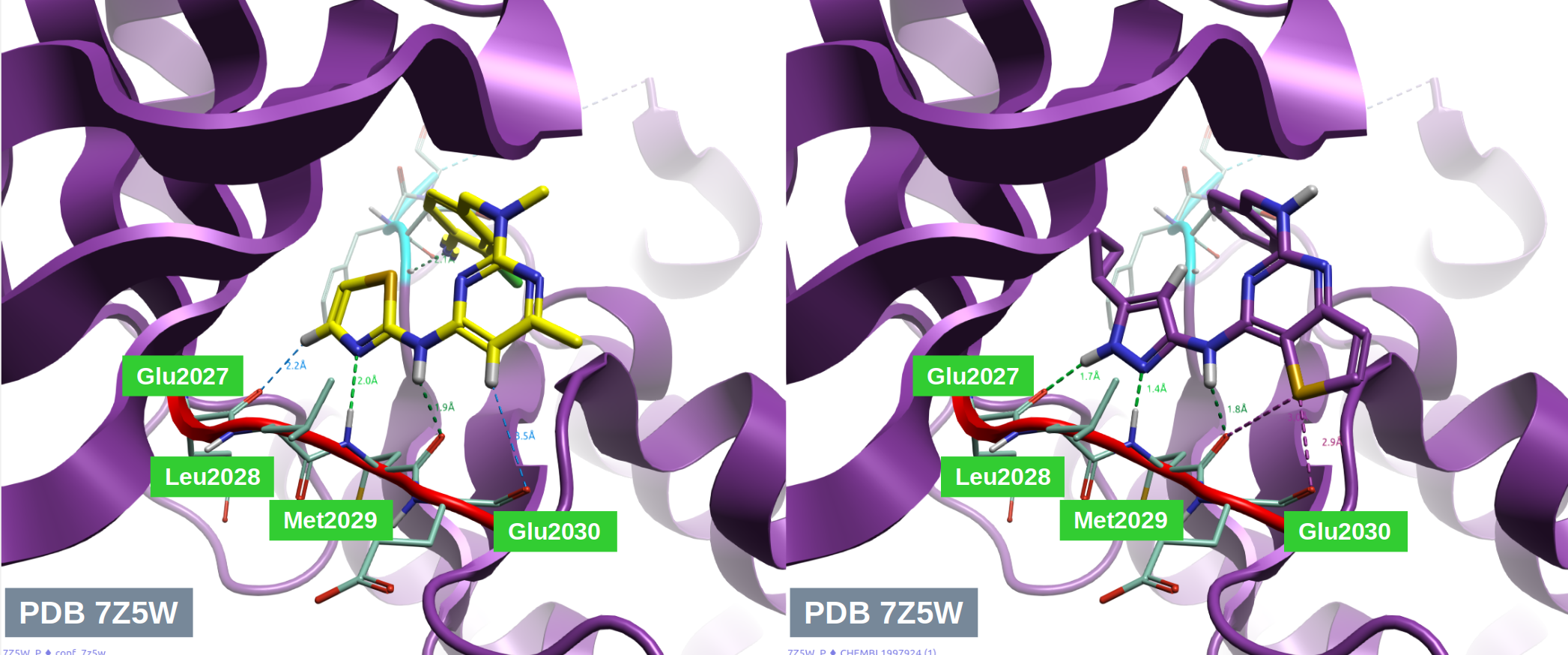
图7. 化合物31活性构象与苗头化合物CHEMBL1997924与ROS1铰链区相互作用的比较。黄色棍棒:化合物31的活性构象CONF_34;紫色棍棒:苗头化合物CHEMBL1997924;紫色飘带:ROS1激酶结构域;红色飘带:激酶铰链区;青色:DFG-motif。
在铰链结合区域,CHEMBL1997924的吡唑-NH-嘧啶并噻吩平面结构与化合物31的噻唑-NH-嘧啶平面结构呈现出完全的空间重叠。值得注意的是,CHEMBL1997924的吡唑环C-H与嘧啶环上的氮原子可形成C-H···N=C分子内氢键,这一特征对于维持其吡唑-NH-嘧啶并噻吩平面结构的稳定性至关重要。如图7所示,CHEMBL1997924的吡唑环NH基团及其氮原子,以及环外连接臂的NH基团,在空间取向上均能与ROS1激酶铰链区形成理想的氢键互补模式。化合物31的嘧啶C-H与Glu2030羰基氧之间形成C-H···O=C氢键相互作用,而在CHEMBL1997924中,相应的相互作用表现为噻吩环硫原子与Glu2030羰基氧之间的O/S超共轭作用(图8)。此外,CHEMBL1997924噻吩环上的硫原子还与Met2029的羰基氧发生O/S超共轭相互作用。
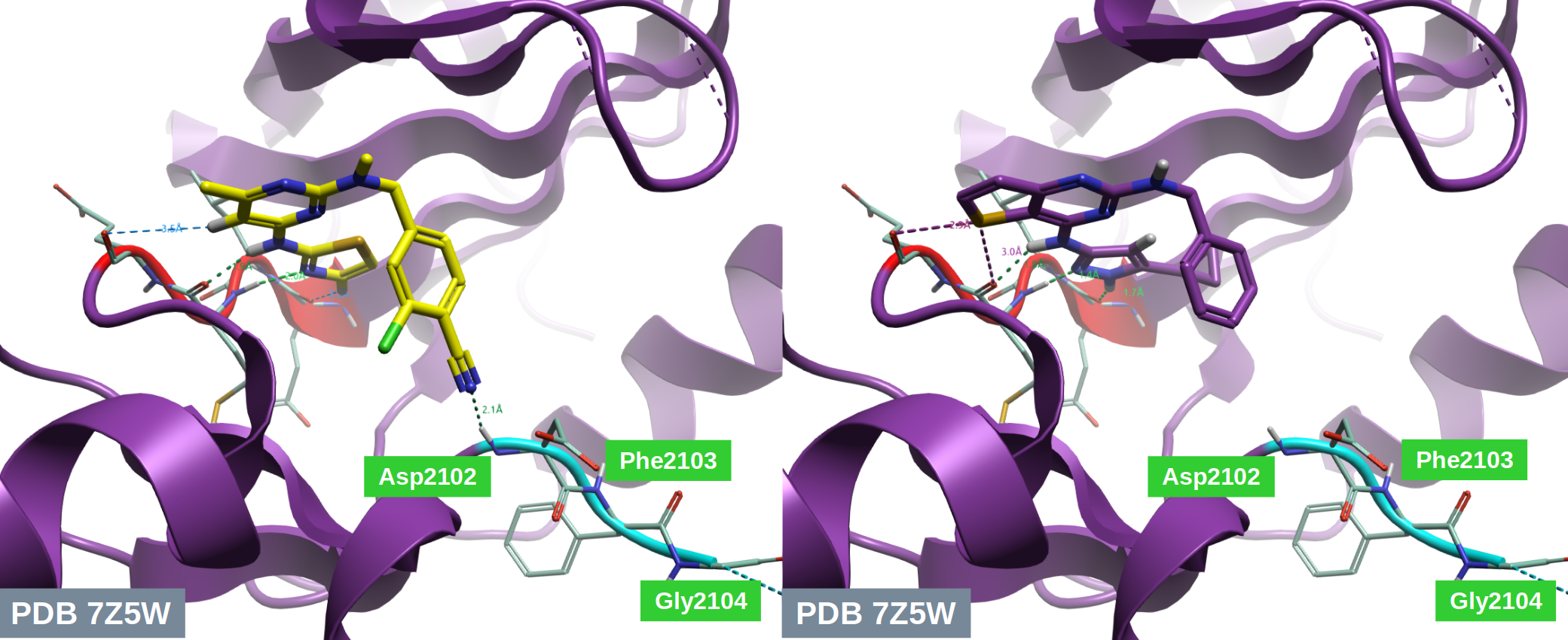
图8.化合物31活性构象与苗头化合物CHEMBL1997924与ROS1 DFG-motif相互作用的比较。黄色棍棒:化合物31的活性构象CONF_34;紫色棍棒:苗头化合物CHEMBL1997924;紫色飘带:ROS1激酶结构域;红色飘带:激酶铰链区;青色:DFG-motif。
在DFG指向区域,如图6右所示,两个分子的苯环取代基不仅空间取向完全一致,且其重原子的空间坐标高度重合, 表明二者具有相似的分子形状特征。然而,与化合物31相比,CHEMBL1997924在苯环对位缺少腈基取代基(图8),因此无法像化合物31那样与DFG区域Asp2102的酰胺NH形成氢键相互作用。
综合构象分析结果表明,CHEMBL1997924与化合物31在分子形状和药效团特征上具有显著相似性,具有与激酶铰链区形成关键相互作用的潜力,这证明了基于查询分子活性构象的虚拟筛选确实召回了具有生物合理性构象的苗头化合物。
2.6 基于非活性构象的虚拟筛选在活性化合物召回方面存在局限性
为评估非活性构象在虚拟筛选中的实际召回能力,有必要对化合物31的非活性构象(CONF_17)与CHEMBL19997924进行构象比对分析。如图9所示,二者在三维空间上呈现出较高的结构匹配度:首先,化合物31的噻唑-NH-嘧啶平面结构与CHEMBL19997924的吡唑-NH-嘧啶并噻吩平面结构呈现显著的空间重叠;其次,两个分子的苯环在空间取向上高度一致;此外,连接臂C-N-C的构象也表现出良好的匹配特性。从分子形状和药效团三维相似性角度分析,该构象匹配确实符合理论要求。
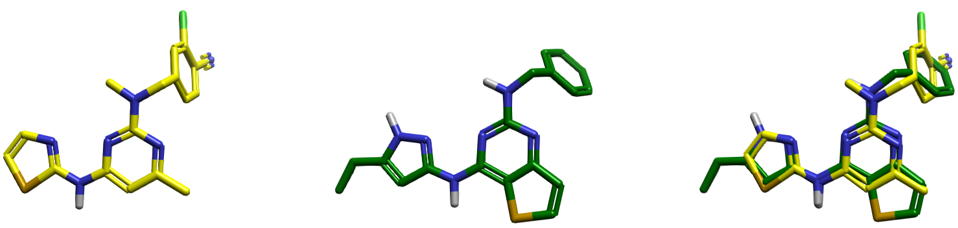
图9. 化合物31的非活性构象与苗头化合物CHEMBL1997924的构象叠合分析。黄色棍状模型:化合物31的非活性构象CONF_17;绿色棍状模型:苗头化合物CHEMBL1997924。
然而,深入分析表明该构象在能量上不可及:吡唑-NH-嘧啶并噻吩共平面排列导致吡唑氮原子与嘧啶氮原子空间相邻,产生显著的电子排斥效应。从构象能垒角度评估,该构象因较高的扭转能垒而处于能量不利状态。更重要的是,在虚拟筛选中,CHEMBL1997924若采用此构象将无法与靶标蛋白活性口袋形成合理的分子间相互作用,因此在严格的后处理筛选流程中必然被排除。综上所述,本案例证明非活性构象在采用严格筛选标准的虚拟筛选中并不具备有效的活性化合物召回能力。
2.7 级联筛选策略对查询结构构象的隐式修正作用
上述结果有助于解释药物发现实践中一个常见的现象:在方法学验证阶段(如使用DUD-E等基准数据集),基于形状或药效团的虚拟筛选方法往往表现出优异的富集能力,能够有效区分已知活性分子(actives)与诱饵分子(decoys);然而,当将相同方法应用于大规模真实化合物库筛选,并辅以严格的多级漏斗式后处理(构象张力能过滤、关键相互作用过滤等)后,最终获得的候选化合物往往数量稀少(其实往往是没有化合物可以保留下来!),且经实验验证大多缺乏显著生物活性。
一个更具迷惑性的观察是:即便在查询分子未经过充分构象优化(例如直接使用2D结构生成的默认3D构象)的情况下,采用“形状/药效团初筛 → 分子对接复筛”的级联虚拟筛选策略,反而常常能获得可验证的活性苗头化合物。这一看似矛盾的现象,实则源于级联流程中第二阶段对接步骤对初始构象偏差的隐式修正作用。具体而言,第一轮基于形状或药效团的筛选本质上是一种高通量的特征匹配过滤器,其主要功能是快速剔除明显不相关的化学空间;而第二轮分子对接不仅重新采样配体在结合口袋中的低能构象,还通过显式的蛋白–配体相互作用评估,自动“纠正”了初始查询构象可能引入的几何或静电偏差。因此,即便初始查询构象非生物活性状态,只要其保留了关键药效团特征,后续对接仍可能识别出能量合理且结合模式正确的构象,并使其在严格的后处理中得以保留。
换言之,级联筛选的成功并非源于第一轮方法的高精度,而在于第二轮对接对构象与结合模式的再优化能力。这也反向验证了本研究的核心结论:查询构象的生物合理性虽非级联流程的绝对前提,但若能在首轮即采用经验证的生物合理性低能构象,将显著提升筛选效率,减少对计算密集型对接步骤的依赖,并降低假阳性率。在资源受限或需极速筛选的场景下(如超大规模库的GPU加速筛选),高质量的查询准备尤为关键。
3. 材料与方法
3.1 查询分子的结构准备
将共晶结构(PDB 3ZBF、4UXL、7Z5X、7Z5W)从蛋白质数据库下载到Flare V10中,并使用Protein | Protein Prep工具小心地准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构分配最佳质子化状态。任何截短的蛋白质链被封端作为蛋白质准备的一部分。全部的共晶结构仅保留A链,并将A链的配体从蛋白中提取出来到配体表单。
使用 Sequence | align 进行序列比对,然后以 3ZBF 为参比,使用 Sequence | superimpose 将另外三个蛋白结构叠合到 3ZBF 上,得到叠合的蛋白结构与配体。
3.2 ChEMBL35的3D数据库准备
ChEMBL35的化合物3D构象系综使用CDPkit的confgen来准备:
1 2 3 4 | confgen -i chembl35.sdf \ -o chembl35_confs.sdf \ -t 60 \ --max-num-out-confs 25 #每个分子最多输出25个构象 |
这个命令暗含了多个默认的参数设置,包括:
- -C MEDIUM_SET_DIVERSE:中等规模多样性集合,采用平衡的构象搜索策略,在计算效率和构象覆盖度之间取得折中
- -c AUTO:程序根据分子特性自动选择系统采样或随机采样
- -e 15:只保留与最低能量构象能量差在15.0 kcal/mol范围内的构象
- -r 0.5:输出构象之间的最小RMSD必须\(≥\)0.5Å,避免结构过于相似
- -S true:忽略输入结构的3D坐标,完全重新生成构象
- -d -q:力场参数设置,系统采样为`MMFF94S_RTOR_NO_ESTAT`; 随机采样为`MMFF94S_RTOR`
3.3 ROSHAMBO2虚拟筛选
ROSHAMBO2虚拟筛选的主要参数如下:
- backend:cuda
- color:true
- start_mode:1
- optim_mode:combination
- max_results:10000
- n_gpus:2
硬件设施为两张Nvidia RTX 4090。总共12分钟完成计算,平均每张卡每秒处理大约19305构象。
3.4 虚拟筛选的统计与排序
见数据部分:result_analysis.py
4. 结论
本研究系统评估了查询分子构象准备策略对基于三维形状的虚拟筛选性能的影响,以阿斯利康发现的ROS1选择性抑制剂化合物31为模型体系。通过结合量子力学优化、共晶结构叠合分析与激酶结合口袋相互作用建模,我们成功识别出一个低能、高相似性且能复现关键蛋白–配体相互作用的生物活性构象(CONF_34)。该构象在铰链区与DFG区均与PDB 7Z5W共晶配体高度一致,验证了其作为可靠查询结构的合理性。
在此基础上,我们构建了一个经过严格标准化与三维构象生成的ChEMBL35衍生数据库(含约135万化合物、2780万构象),并采用ROSHAMBO2平台分别以活性构象(CONF_34)与高能非活性构象(CONF_17)作为查询结构进行大规模虚拟筛选。尽管两种构象在整体评分分布上无显著差异,活性构象在已知ROS1抑制剂的召回能力与排序优先级方面表现出明显优势。具体而言,高亲和力苗头化合物CHEMBL1997924(Ki = 1 nM)在活性构象筛选中以RefTverskyCombo评分为1.88位列第11位(标准竞争排名),而在非活性构象下仅排第50位(评分为1.81);若采用传统Tanimoto Combo评分,其排名差距进一步扩大至52 vs. 299。这一结果凸显了查询构象的生物相关性对虚拟筛选排序性能的关键影响。
值得注意的是,尽管非活性构象(CONF_17)在几何上能与部分活性分子实现局部形状匹配(如图7所示),但其高构象能垒(8.5 kcal/mol)及与靶标结合模式的不兼容性,使其在实际药物发现流程中难以有效引导活性化合物的富集。该现象揭示了一个重要原则:仅依赖三维形状重叠而忽视构象能量合理性与结合模式一致性的虚拟筛选策略,可能导致假阳性命中或活性分子的低效召回。
此外,本研究还验证了Tversky组合评分(尤其是RefTverskyCombo,α=0.95)在本案例中优于传统Tanimoto组合评分的排序能力,这与文献中关于“偏向查询分子”的相似性度量在先导化合物识别中更具敏感性的观点一致。这一发现为后续基于形状的虚拟筛选参数优化提供了实证依据。
综上所述,查询分子的构象准备不仅是技术性预处理步骤,更是决定虚拟筛选成败的核心环节。推荐在实际应用中优先采用基于生物活性相关的低能构象作为查询结构,并结合Tversky类评分函数以提升活性化合物的早期识别效率。
5. 数据
虚拟筛选的查询分子与结果文件可从GITHUB数据库仓库gkxiao/RoshamboLearningJourney的data目录下载:
- 化合物31的构象文件: 31_CONFS_QM.sdf
- 虚拟筛选统计、排序分析:result_analysis.py
- CONF_17: query_CONF_17.sdf
- CONF_34: query_CONF_34.sdf
- CONF_17虚拟筛选结果: CONF_17_VS_hits.sdf.gz, CONF_17_VS_hits_score.csv
- CONF_34虚拟筛选结果: CONF_34_VS_hits.sdf.gz, CONF_34_VS_hits_score.csv
- 化学基因组分析(靶标预测)报告: target_prediction_report.md
注意:这里的CONF_17不是31_CONFS_QM.sdf里的第17号构象CONF_17!
6. 推荐阅读
如何在没有蛋白共晶结构的情况下预测生物活性相关构象:bioactive conformation
7. 接下来可以做什么
ROSHAMBO作为分子相似性搜索工具,比起Blaze与ROCS等商业软件,其局限性在于无法对查询式(query)施加约束条件或进行编辑操作,例如对分子形状或药效团特征设置权重等。这种限制导致高评分化合物不一定包含与靶标形成关键相互作用所需的特征。因此,采用级联二次筛选策略显得尤为重要。例如,在Flare软件环境中,可对查询分子设置药效团或场点约束,随后利用XED场相似性进行重新评分,以确保筛选出的化合物满足包含关键相互作用的药效团特征。
ChEMBL作为化学基因组学数据库,其应用基于”相似性质原理”(similarity property principle)的假设,即三维结构相似的化合物倾向于表现出与相同的结合位点相互作用与相似的生物学活性。基于此原理,计算结果可直接用于预测化合物的潜在治疗靶标(on-target)与脱靶(off-target)。关于化合物31的靶标预测与反靶预测,另外写了一篇博客文章进行介绍,请点击这里:化合物31的化学基因组学分析。
ROSHAMBO2可作为插件集成到Vida或Flare分子建模软件平台中,作为初级虚拟筛选工具使用。这种集成方式不仅扩展了原有软件的功能,还为研究人员提供了一种高效的化合物筛选解决方案,特别适用于药物发现早期阶段的化合物库筛选。
8. 文献
- Petrović, D. et al. (2022) “Virtual Screening in the Cloud Identifies Potent and Selective ROS1 Kinase Inhibitors,” Journal of Chemical Information and Modeling, 62(16), pp. 3832–3843. Available at: https://doi.org/10.1021/acs.jcim.2c00644.
- FastROCS. GPU-accelerated Shape Similarity Search. Openeye, Cadence Molecular Sciences. https://www.eyesopen.com/fastrocs
- Nicholls, A. et al. (2010) “Molecular Shape and Medicinal Chemistry: A Perspective,” Journal of Medicinal Chemistry, 53(10), pp. 3862–3886. Available at: https://doi.org/10.1021/jm900818s.
- A similarity search was conducted on October 9, 2025, using the chemical structure “Cc1cc(Nc2nccs2)nc(N(C)Cc2ccc(C#N)c(Cl)c2)n1” as the query molecule with a similarity threshold of 40% via the ChEMBL database (https://www.ebi.ac.uk/chembl/advanced_search/similarity/Cc1cc(Nc2nccs2)nc(N(C)Cc2ccc(C%23N)c(Cl)c2)n1/40). No compounds were retrieved under these search conditions.
- ROCS. Openeye, Cadence Molecular Sciences. https://www.eyesopen.com/rocs
- https://github.com/gkxiao/ROCS-Query-Prep-Benchmark
- https://www.ebi.ac.uk/chembl/explore/compound/CHEMBL35
- CDPKit (Version 1.2.3). https://github.com/molinfo-vienna/CDPKit
- Seidel, T. et al. (2023) “High-Quality Conformer Generation with CONFORGE: Algorithm and Performance Assessment,” Journal of Chemical Information and Modeling, 63(17), pp. 5549–5570. Available at: https://doi.org/10.1021/acs.jcim.3c00563.
- Atwi, R. et al. (2025) “ROSHAMBO2: Accelerating Molecular Alignment for Large Chemical Libraries with GPU Optimization and Algorithmic Advances,” Journal of Chemical Information and Modeling [Preprint]. Available at: https://doi.org/10.1021/acs.jcim.5c01322.
- Atwi, R. et al. (2024) “ROSHAMBO: Open-Source Molecular Alignment and 3D Similarity Scoring,” Journal of Chemical Information and Modeling, 64(21), pp. 8098–8104. Available at: https://doi.org/10.1021/acs.jcim.4c01225.
- Horvath, D., Marcou, G. and Varnek, A. (2013) “Do Not Hesitate to Use Tversky—and Other Hints for Successful Active Analogue Searches with Feature Count Descriptors,” Journal of Chemical Information and Modeling, 53(7), pp. 1543–1562. Available at: https://doi.org/10.1021/ci400106g.
- 肖高铿. 在基于配体3D相似性的虚拟筛选中使用Tversky. 墨灵格的博客. http://blog.molcalx.com.cn/2023/04/17/using-tversky-in-vs.html
- Fischer, A. et al. (2021) “Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results,” Journal of Medicinal Chemistry, 64(5), pp. 2489–2500. Available at: https://doi.org/10.1021/acs.jmedchem.0c02227.


