
摘要:自分泌运动因子 (ATX) 抑制剂 GLPG1690 的临床开发虽终止,但其先导化合物优化过程具有重要的方法学借鉴意义。针对该项目中传统基于结构药物设计 (SBDD) 未能指导关键腈基取代的问题,本文采用回溯性分析,结合分子动力学与网格非均匀溶剂化理论 (GIST) 进行研究。结果表明,经典分子对接无法捕捉腈基引入带来的活性变化。apo-GIST 分析成功识别出噻唑环附近的高能水合位点,配体置换自由能计算定量预测了结合增益。然而,研究亦揭示单一静态结构的局限性:水置换收益并非腈基专属,且初始结构 (PDB 5M7M) 中的静电排斥可能误导设计优先级,反映了难以预见诱导契合效应的“静态结构陷阱”。本研究建议,在水热力学分析识别关键位点的同时,从业人员需时刻警惕静态结构的潜在缺陷,结合蛋白构象柔性评估与化学特性分析,以克服单一结构信息的误导,实现更精准的理性药物设计。
肖高铿/2021-08-24:第一次发布
肖高铿/2026-02-17:补充了水密度、水合位点、溶剂化自由能、配体置换自由能
Gaokeng Xiao. 2026-02-17. The Trap of Static Structures and Insights from Water Thermodynamics: A Retrospective Study on the Lead Optimization of ATX Inhibitor GLPG1690.
1. 前言
特发性肺纤维化(Idiopathic Pulmonary Fibrosis, IPF)是一种预后较差的慢性肺部疾病,自分泌运动因子(Autotaxin, ATX)作为溶血磷脂酸(LPA)生成的关键酶,是 IPF 治疗的重要靶点。Galapagos 公司针对该靶点开发了首创抑制剂 GLPG1690(图1化合物 2)。尽管该化合物在早期临床试验中显示出积极成果,但最终因 III 期临床试验无效而终止开发。虽然临床开发失败,但其从先导化合物到候选药物的优化过程却是计算机辅助药物设计(CADD)的典型案例。
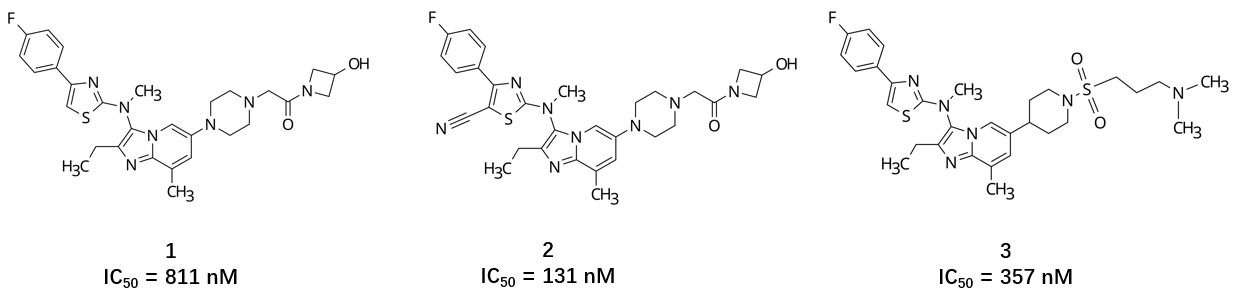
图1. ATX 抑制剂 1、2、3的化学结构式。
图 1 亦概括了 Galapagos 公司 ATX 项目的优化历史1,2。在研发早期,Galapagos 解析了化合物 3 与 ATX 的复合物晶体结构(PDB 5M7M),但这一结构信息并未直接转化为高活性化合物的理性设计1。在项目前半阶段,团队共合成并测试了 157 个化合物,主要针对噻唑环进行了 6 种取代基修饰。值得注意的是,该阶段未采用溶剂化分析工具,首个具有潜力的噻唑环腈基取代衍生物是通过传统配体优化策略发现的(如图 2 所示)。
图2. ATX 抑制剂骨架及提高活性的腈基取代位置。回溯性分析显示,共计 27 个引入腈基的设计实例均观察到活性提升,无反例。
如图 3 项目时间线所示,在确认引入腈基可显著提升活性(平均 pIC50 提升约 0.8 个对数单位)后,约 1/3 的后续化合物在噻唑环或相邻苯环邻位引入了腈基取代。最终进入临床研究的化合物 2 亦受此策略影响。
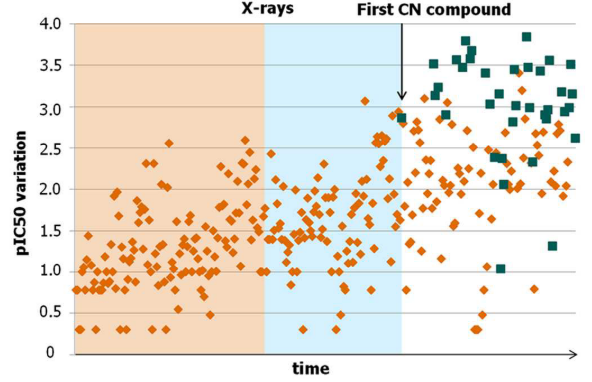
图3. 项目发展时间线。每个数据点代表一个经生化活性测试的化合物。深黄色:无腈基化合物;墨绿色:含腈基化合物。黄色区域:ATX-抑制剂共晶结构解析前的化合物;蓝色区域:共晶结构解析后、但在通过传统方法发现首个腈基取代物之前合成测试的化合物;白色区域:发现首个腈基化合物后合成测试的化合物。图片来源:文献 1。
尽管已获取化合物 3 与 ATX 的 X-射线衍射晶体结构(PDB 5M7M,细节见文献 2),但仅基于静态结构进行优化面临挑战。图 4A 显示,该系列化合物存在多个理论上可行的取代生长方向(红色箭头),针对噻唑环 5 位进行取代修饰并非显而易见的策略。仔细观察配体周围蛋白表面发现,噻唑附近存在一个足以容纳水分子的空腔(图 4B),但在 X-衍射结构的电子密度图中并未观测到该位置存在水分子。尽管如此,结合口袋上方确实存在一个稳定的结晶水分子(图 4C)。使用分子对接软件 Glide 进行的计算表明,其打分函数未能预测腈基引入带来的活性增益。在本案例中,腈基取代化合物是通过传统配体优化发现的。项目完成后的分子动力学(MD)模拟回溯性分析表明,该空腔可被水分子瞬时(transiently)占据,且该水分子无法与周围蛋白残基形成稳定的氢键网络(图 4D)。这种不稳定的水分子存在,为解释腈基对化合物活性的贡献提供了理论依据。
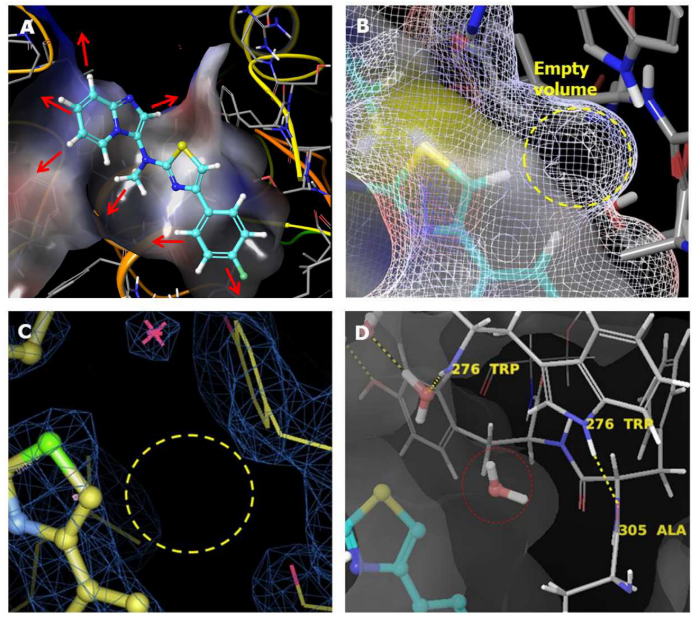
图4. (A)ATX 结合位点表面。红色箭头指示 7 个不与蛋白发生立体碰撞的生长方向,噻唑环 5 位取代并非显而易见。(B)精细蛋白表面网格显示噻唑附近存在可容纳水分子的空腔。(C)共晶结构电子密度图(2Fo-Fc@1σ)未显示噻唑 5 位附近存在水分子。(D)50 ns 分子动力学模拟表明噻唑 5 位附近存在瞬态水合位点,但该水分子与邻近蛋白残基无氢键作用。图片来源:文献 1。
2. 研究目的
本文旨在通过回溯性分析,探讨以下关键科学问题:
- 在获取首个 X-射线衍射数据后,能否利用计算方法将先导化合物优化方向从图 4-A 所示的众多可能性中,聚焦并引导至噻唑环修饰?
- 若确定对噻唑环进行修饰,基于结构的设计(SBDD)能否提出引入腈基的具体方案?
- 如何从蛋白 – 配体相互作用角度理解构效关系(SAR),解释图 1 中化合物 1 与 2 的活性差异?
3. 计算方法
3.1 蛋白的结构准备
从蛋白质数据库(PDB)下载配体 – 蛋白质复合物结构(PDB ID: 5M7M 和 5MHP)至 FlareTM (Version 11)软件3中。使用 Protein Prep 工具进行标准化预处理,包括添加氢原子、优化氢键网络、消除原子冲突及分配最佳质子化状态。截短的蛋白质链末端进行了封端处理。使用多重序列比对工具在 Flare 中比对蛋白序列,随后通过 Cα 原子的最小二乘法拟合进行结构叠合。
3.2 apo-GIST分析
GIST4 是一种使用显式水对蛋白质进行约束的分子动力学模拟,并使用非均匀溶剂化理论 (inhomogeneous solvation theory,IST) 计算预定义体积(通常是结合位点)的水分子分布和热力学性质。在本文中,用Flare 实现的GIST方法5分别对 PDB 5M7M 和 5MHP 结合位点进行了无配体的GIST分析,使用了如下条件:
- Calculation method: Normal
- Ligand: None
- Grid spacing:0.5 Å
- Grid Definition:Ligand
- Chains: A, Water
- Simulation length: 20ns
- Solvent Model: explicit TIP3P Water
3.3 蛋白相互作用势(PIP)分析
蛋白相互作用势(Protein Interaction Potential, PIP)是 Cresset 分子相互作用势在蛋白体系上的延伸,均基于 XED 力场计算。该方法原理类似于配体场计算:用探针原子填充蛋白活性位点,计算每个格点上的相互作用势6,7。为更好地处理蛋白结构中大量带电基团,该方法采用了 Mehler 等人8提出的距离依赖介电函数。为解析化合物 1 与 2 的活性差异,在 Flare 中对 PDB 5M7M 与 5MHP 的结合位点进行了 PIP 分析。
3.4 配体场分析
基于化合物 2 与 ATX 复合物结构(PDB 5MHP),在 Flare 中对化合物 2 进行结构编辑(删除腈基或替换为甲基等)以模拟化合物 1 的结合模式,随后计算化合物 1 和 2 的配体场6,7。
4. 结果
4.1 化合物 1、2 与 ATX 的相互作用及分子对接打分比较
在项目开发期间,解析了化合物 3(见图 1)与 ATX 的共晶结构 PDB 5M7M(图5 左)。由于噻唑环附近空腔体积相对较小,该区域未被视为优先优化位点。随后解析的化合物 2 共晶结构 PDB 5MHP(图5 右)显示,化合物 2 的腈基填充了该空隙。
从 5MHP 复合物结构分析,未观察到腈基与蛋白之间存在经典的氢键或静电相互作用。通过在 Flare 中编辑 5MHP 配体(删除腈基)构建化合物 1 的结合模式,并使用分子对接软件 GNINA9 的 Vina Score10 对化合物 1 与 2 进行打分。结果如表 1 所示,预测的结合自由能无显著差异,分别为 -11.046 与 -11.149 kcal/mol(见表 1 Affinity)。这表明代表性分子对接方法(Vina score)无法区分化合物 1 与 2 的活性差异,这与 Bucher 等人1 报道的 Glide 对接结果一致。即使采用 GNINA 内置的深度学习打分函数 CNNaffinity(见表 1),亦未能有效区分两者。
| Compound | 1 | 2 |
|---|---|---|
| gauss_1 | 105.70258 | 118.43863 |
| gauss_2 | 2107.71216 | 2235.38745 |
| repulsion | 1.74530 | 2.12076 |
| hydrophobic | 68.58022 | 68.58022 |
| non_dir_h_bond | 0 | 0 |
| Affinity (kcal/mol) | -11.046 | -11.149 |
| CNNaffinity | 8.580 | 8.728 |
| Exp ΔG (kcal/mol) | -8.30 | -9.38 |
计算基于PDB 5MHP
此外,化合物 1 与 2 对接打分中的氢键相互作用项贡献均为 0,这与 Bucher 等人1 未观察到腈基参与氢键相互作用的结果一致。综上所述,经典的分子间相互作用分析及主流分子对接打分(Vina、Glide)均难以区分化合物 1、2 的活性差异,亦无法阐明腈基在相互作用中的具体角色,这表明在先导化合物优化中,仅依赖传统对接打分指导新分子设计与排序存在局限性。
4.2 化合物 2、3 结合模式及蛋白构象变化比较
比较化合物 2、3 与 ATX 的共晶结构(PDB 5M7M 与 5MHP),如图 5 所示,发现噻唑环 C-H 指向的 Loop 区域存在显著差异,尤其是残基 Gly272-Thr273-Phe274(紫红色部分)的空间取向截然不同。
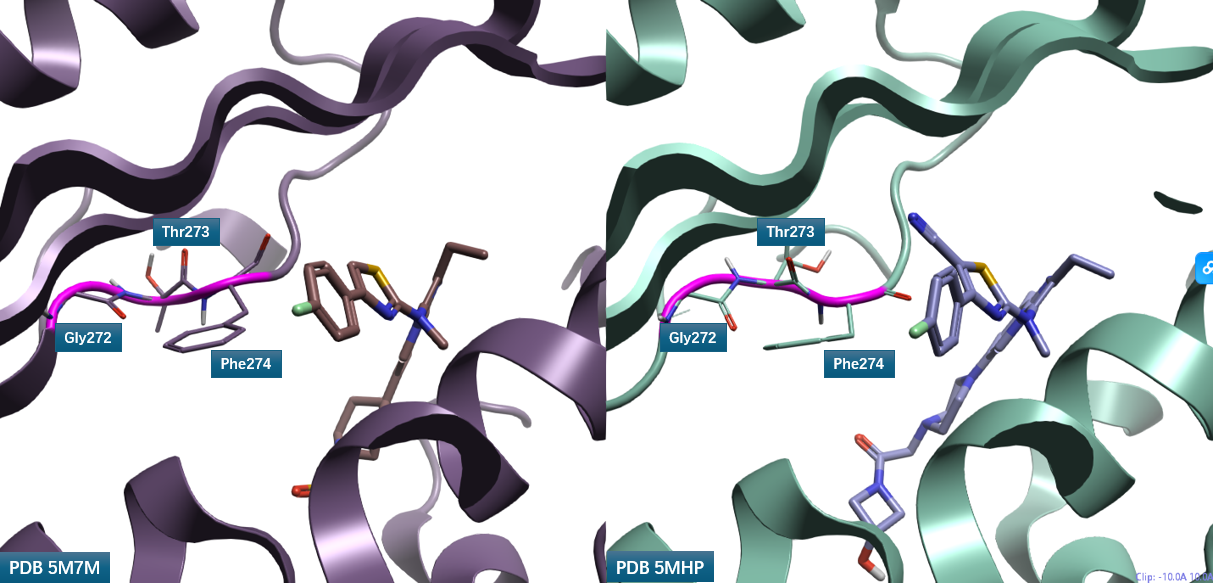
图5. 化合物2(右)、3(左)与 ATX (飘带)共晶结构PDB 5MHP、5M7M的结合位点比较
PDB 5M7M 与 5MHP 中 Loop 残基 Gly272-Thr273-Phe274 的构象差异巨大(图 6)。在 5M7M 中,Phe274 的羰基氧靠近化合物 3 的噻唑环位置;而在 5MHP 中,Phe274 的羰基氧远离化合物 2 的腈基。这种残基取向差异导致化合物 2 腈基所在位点(红色圆圈高亮)的蛋白静电场性质截然相反:5M7M 在该位置呈现负静电场,与腈基氮原子的负静电场排斥,而与噻唑 C-H 配体场互补;5MHP 则呈现正蛋白相互作用势(PIP),与腈基氮原子的负静电场互补,而与噻唑 C-H 配体场排斥。
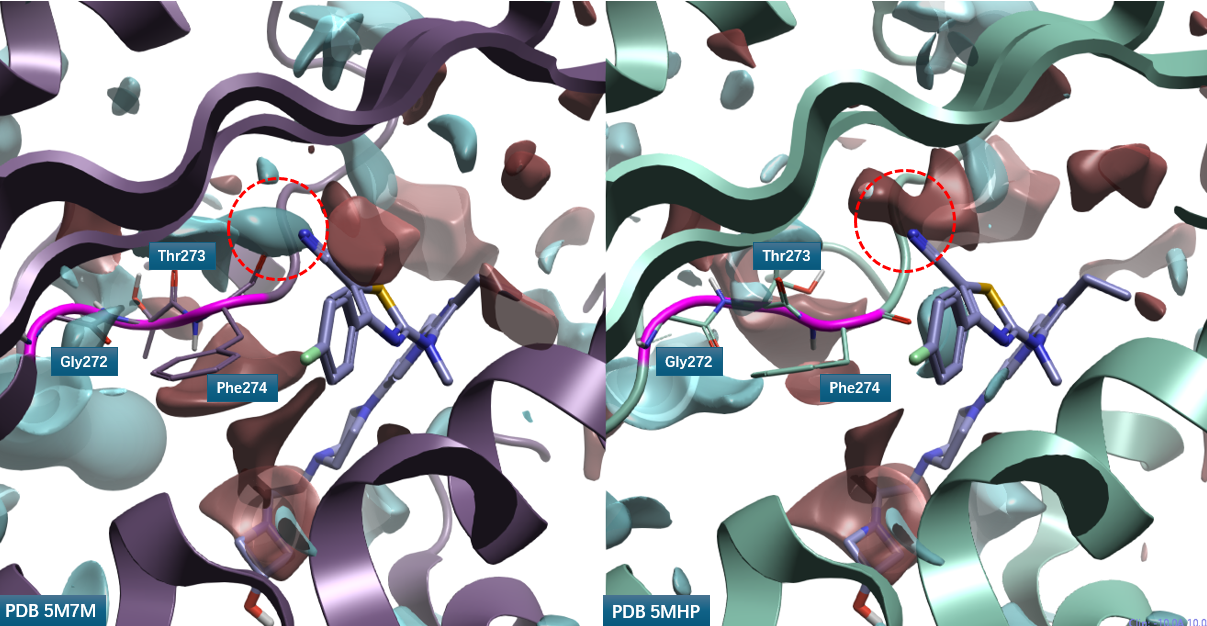
图6. 化合物2、3与 ATX (飘带)共晶结构PDB 5MHP、5M7M的PIP比较
图 7 展示了化合物 1、2 的配体场。化合物 1 中噻唑 C-H 方向呈现红色正配体场,与 PDB 5M7M 该方向的负 PIP 互补;化合物 2 中,腈基附近的蓝色负配体场与 PDB 5MHP 的正 PIP 互补。
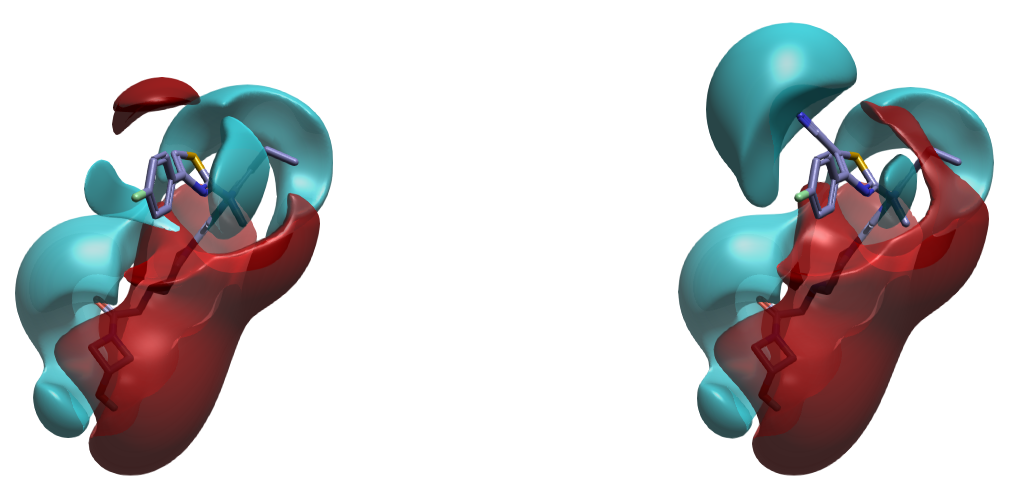
图7. 化合物1、2配体场
综上,在化合物 1 噻唑 C-H 上引入腈基会诱导 ATX 蛋白发生轻微构象变化,进而导致该方向蛋白相互作用场(PIP)的反转,最终实现静电互补。然而,仅从化合物 3 与 ATX 共晶结构(PDB 5M7M)出发,由于难以预见 Loop 构象变化引起的 PIP 反转,因此无法单纯依据 PIP 分析指导在噻唑 C-H(5-位)上引入腈基的设计,甚至因为静电冲突而降低引入腈基的优先级。
4.3 结合口袋的apo-GIST分析
对化合物3与ATX共晶结构PDB 5M7M进行apo-GIST分析,图8展示了水密度(Water Density =4)与水合自由能ΔG在化合物3苯基噻唑片段占据口袋的分布。在靠近噻唑C-H方向上的高密度且红色高能的水分布,确实对该位置引入新的基团起到足够的提示作用。
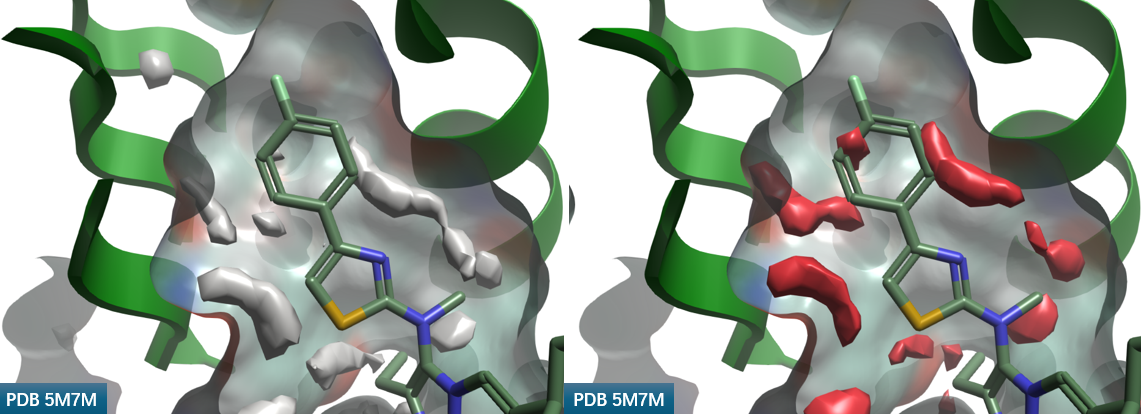
图8. PDB 5M7M的apo-GIST分析结果。灰色等值图:\(Water\ Density\ =\ 4\) ,红色等值图:\(\Delta G \ge 0.5 kcal/mol\);绿色等值图:\(\Delta G\ \le\ -0.5\ kcal/mol\)
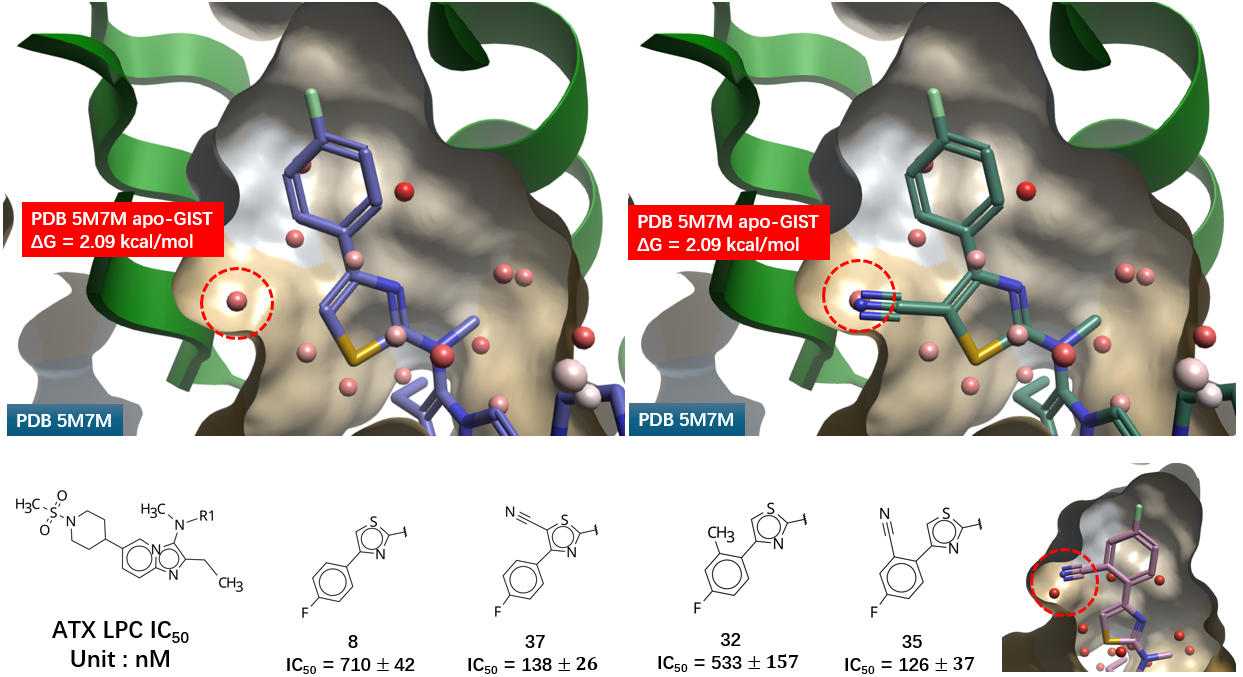
图 9 进一步展示了通过峰值拾取法聚类得到的水合位点。噻唑 C-H 方向上的水合位点(红色圆圈高亮)位于疏水性子口袋内,其溶剂化自由能ΔG 高达 2.09 kcal/mol,直接提示了通过引入基团置换该高能水分子以提升活性的机会。
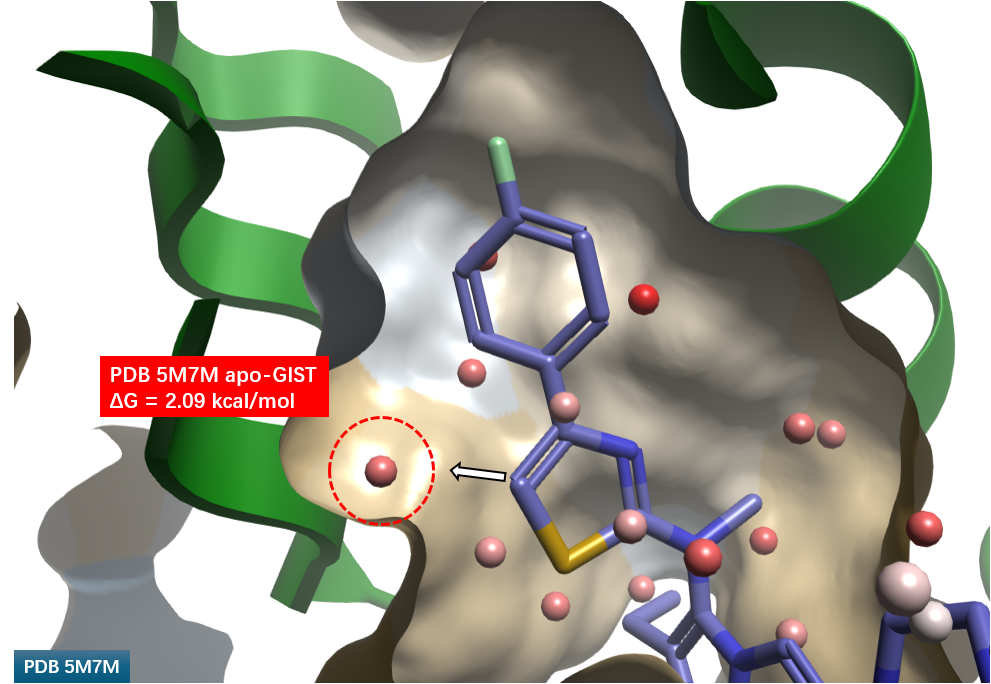
图9. 基于apo-GIST的水合位点。飘带:ATX;分子表面:疏水性着色;棍棒:化合物3. 球体:水合位点。
在 Flare 中对化合物 3 进行编辑,于噻唑 C-H 位引入腈基。结果显示腈基位置与疏水口袋中的高能水合位点高度重合(图 10 红色圆圈高亮),意味着实现了对高能水分子(ΔG = 2.09 kcal/mol)的完美置换。
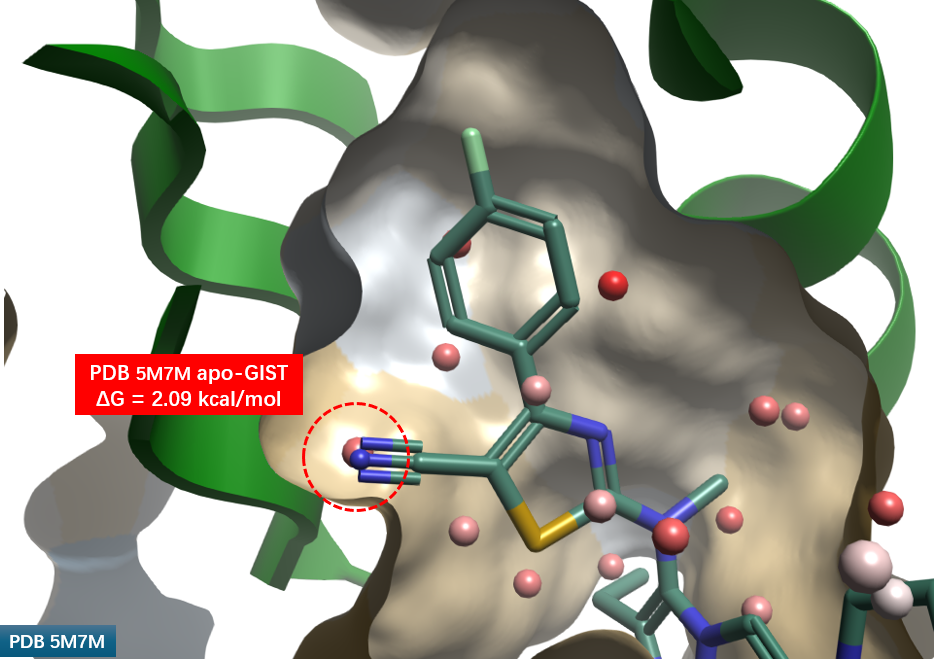
图10. PDB 5M7M apo-GIST水合位点的置换。飘带:ATX;分子表面:疏水性着色;棍棒:化合物2. 球形分子:水合位点。
综上所述,通过 apo-GIST 分析的水密度图、ΔG 等值面及水合位点分析,确实能够将优化目标聚焦至少数关键修饰位点,尤其是邻近噻唑环及苯环的高能水合位点,为药物化学家提供了明确的方向性指导。
4.4 配体置换自由能 \(ΔG_{watdisp}\)
当配体原子占据原本由水分子占据的结合位点区域时,将引起局部去溶剂化效应,从而产生与水置换相关的自由能变化,记为 \(ΔG_{watdisp}\)。该量通过累积配体原子范德华半径范围内体素的 GIST 热力学量,并进行体积积分计算:
$$
\Delta G_{watdisp} = \sum_{i \in {ligand}}\Delta g_{i}⋅V_{voxel}\cdots(1)
$$
其中\(Δg_i\)为体素 i 的自由能密度(kcal/mol/ų),\(V_{voxel}\)=0.125 ų。计算通过pyflare脚本实现。
| Compound | 3 | 3-CN |
|---|---|---|
| ΔGwatdisp (kcal/mol) | -12.291 | -14.095 |
| Exp ΔG (kcal/mol) | -8.38 | -9.35 |
计算基于PDB 5M7M
基于 PDB 5M7M 的 Apo-GIST 分析及公式 1 计算,化合物 3 及其腈基取代衍生物的配体置换自由能如表 2 所示,分别为 -12.291 和 -14.095 kcal/mol。这意味着在噻唑 C-H 上引入腈基后,可获得 -1.80 kcal/mol 的水分子置换自由能增益(ΔΔGwatdisp)。引入腈基的同时也增加配体的去溶剂自由能以及诱导蛋白构象变化,这些可能都对结合不利,实际的结合自由能增益并没有预期那么大。无论如何,尽管计算的自由能增益高于实验测得的ΔΔG (0.97 kcal/mol),但确实定量预测了引入腈基有利于结合的趋势。
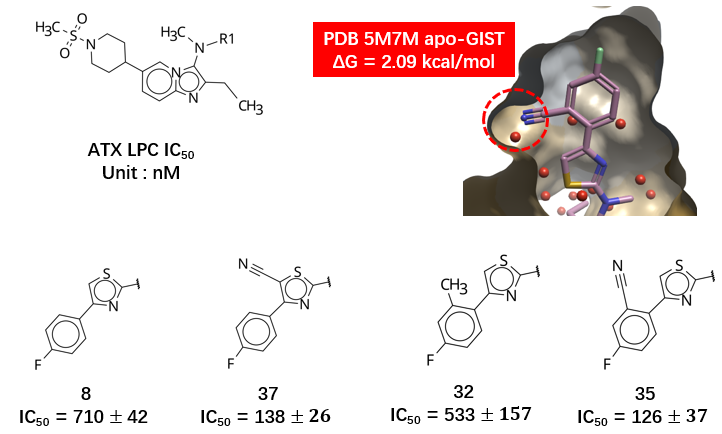
图11. ATX抑制剂对PDB 5M7M apo-GIST水合位点的置换。飘带:ATX;分子表面:疏水性着色. 球形分子:水合位点。
然而,需指出的是,配体置换自由能的增益并非腈基所独有:任何能够有效占据该高能水合位点且空间位阻适宜的基团,理论上都可获得类似的热力学收益。噻唑5-位也非置换同一高能水合位点的唯一取代位置。如图11的分子对2所示,无论是噻唑环、还是苯环上的取代都可能因高能水分子置换而提高活性。此外,结合 PIP 分析结果(见图 7),在 PDB 5M7M 的静态构象中,噻唑 5 位附近呈现负静电势,与腈基氮原子的负电性存在静电排斥,这在一定程度上会降低引入腈基的设计优先级。因此,若仅依赖 5M7M 单一静态结构进行理性设计,既难以从热力学角度特异性识别腈基的优势,又可能因静电冲突的误判而低估该修饰策略的潜力。这一结果进一步凸显了结合动态水热力学分析与多构象蛋白场评估在先导化合物优化中的必要性。
5. 结论
Bucher 等人1,2 在 ATX 抑制剂先导化合物优化过程中指出,尽管解析了抑制剂 -ATX 的复合物结构,但传统的相互作用分析及经典 Glide 分子对接打分未能为药物化学家提供优先修饰噻唑环的提示,导致项目不得不依赖传统试错法发现腈基取代方案。即便在获得首个腈基取代化合物后,传统方法仍难以正确解释 SAR,限制了项目理解与进度。该案例提出了三个核心问题:(1)在获得首个共晶结构后,如何从众多策略中识别出噻唑环修饰的高优先级?(2)确定修饰位点后,如何理性引入腈基?(3)如何从机理上理解噻唑环的 SAR?
本文通过回溯性研究尝试回答上述问题。分子对接打分(GNINA/Vina)结果显示,主流的对接方法无法区分引入腈基带来的活性提升。对比化合物 2、3 的共晶结构发现,腈基引入诱导了蛋白 Loop 构象变化,进而改变蛋白相互作用场(PIP),这是传统对接方法无法捕捉的,也解释了为何不能单独利用静态结构的 PIP 给出引入腈基的建议。
我们进一步采用 apo-GIST 对共晶结构进行分析,通过水密度、ΔG 及水合位点的可视化,成功将优化目标聚焦至噻唑 C-H 方向的子口袋。然而,本研究也揭示了单一方法的局限性:首先,水分子置换带来的自由能增益并非腈基所独有,任何能占据该高能位点的基团理论上均可获益,GIST 本身无法特异性指向腈基;其次,基于 PDB 5M7M 静态结构的 PIP 分析显示该位点存在负静电势,与腈基产生静电排斥,若仅依赖该静态结构,反而可能降低引入腈基的优先级。
综上所述,本研究表明,在静态结构信息不足以指导设计时,结合水热力学分析(如 GIST)能有效识别高能水分子位点,提供关键的热力学驱动力依据。但必须深刻认识到,在真实药物研发场景中,当仅拥有初始静态结构(如 PDB 5M7M)时,研究人员往往难以预见配体诱导的蛋白构象变化(Induced Fit)。这构成了基于结构药物设计的一个潜在“陷阱”。静态结构永远存在潜在的缺陷,其展示的静电环境或空间位阻可能仅代表了某一亚稳态,而非最终结合态,极易误导设计决策。 因此,从业人员需时刻警惕单一静态结构的局限性,在设计决策中应充分考量蛋白柔性带来的不确定性,将水热力学分析与多构象系综评估及化学直觉相结合,方能克服静态结构的固有缺陷,为先导化合物优化提供更稳健的决策支持。
6. 数据
PDB 5M7M的apo-GIST计算结果的Flare项目文件:Autotaxin。如果你还不是Flare的用户,可以申请一个免费的Flare Viewer来查看Flare的flr项目文件。
7. 想在自己的项目中使用GIST,联系我们,获取软件试用
想要尝试Flare信息丰富、用户友好界面,发现它如何帮助您自信地推动潜在先导化合物优化?请现在就联系我们安排试用,快速访问Flare的广泛功能。我们的专业团队随时准备通过安装和设置为您提供支持,而我们全面的教程库——涵盖从常见工作流程到高级方法和功能的所有内容将帮助您开始使用。我们在这里帮助您更快地实现目标,让您设计出感兴趣的分子。
电邮:info@molcalx.com
电话:020 – 38261356
8. 文献
- Bucher, D., Stouten, P., & Triballeau, N. (2018). Shedding Light on Important Waters for Drug Design: Simulations versus Grid-Based Methods. Journal of Chemical Information and Modeling, 58(3), 692–699. https://doi.org/10.1021/acs.jcim.7b00642
- Joncour, A., Desroy, N., Housseman, C., Bock, X., Bienvenu, N., Cherel, L., Labeguere, V., Peixoto, C., Annoot, D., Lepissier, L., Heiermann, J., Hengeveld, W. J., Pilzak, G., Monjardet, A., Wakselman, E., Roncoroni, V., le Tallec, S., Galien, R., David, C., … Heckmann, B. (2017). Discovery, Structure–Activity Relationship, and Binding Mode of an Imidazo[1,2- a ]pyridine Series of Autotaxin Inhibitors. Journal of Medicinal Chemistry, 60(17), 7371–7392. https://doi.org/10.1021/acs.jmedchem.7b00647
- Flare (Version 11). https://cresset-group.com/software/flare
- Nguyen, C.N., Kurtzman Young, T. and Gilson, M.K. (2012) “Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril,” Journal of Chemical Physics, 137(4), pp. 973–980. Available at: https://doi.org/10.1063/1.4733951.
- Flare GIST. https://cresset-group.com/software/flare-3drism
- Cheeseright, T., Mackey, M., Rose, S., & Vinter, A. (2006). Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. Journal of Chemical Information and Modeling, 46(2), 665–676. https://doi.org/10.1021/ci050357s
- Bauer, M. R., & Mackey, M. D. (2019). Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. Journal of Medicinal Chemistry, 62(6), 3036–3050. https://doi.org/10.1021/acs.jmedchem.8b01925
- Mehler, E. L. (1996). Self-Consistent, Free Energy Based Approximation To Calculate pH Dependent Electrostatic Effects in Proteins. The Journal of Physical Chemistry, 100(39), 16006–16018. https://doi.org/10.1021/jp9537926
- McNutt, A. T., Francoeur, P., Aggarwal, R., Masuda, T., Meli, R., Ragoza, M., Sunseri, J., & Koes, D. R. (2021). GNINA 1.0: molecular docking with deep learning. Journal of Cheminformatics, 13(1), 43. https://doi.org/10.1186/s13321-021-00522-2
- Trott, O.; Olson, A. J. AutoDock Vina. J. Comput. Chem. 2010, 31, 445–461. https://doi.org/10.1002/jcc.21334.
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, acs.jcim.1c00203. https://doi.org/10.1021/acs.jcim.1c00203.


