
摘要:Schulz R等人(2018 JMC)采用基于结构的方法识别了3C蛋白酶(3C Protease)共价键结合的3D药效团模型;并用包含33个活性化合物、1771个decoy化合物的数据集对该药效团模型进行验证,发现该模型具有非常好的敏感性与专一性;对一个3000个化合物的数据库进行虚拟筛选发现3C蛋白酶共价键结合的不可逆抑制剂,最后通过骨架跃迁、结构优化发现了化学稳定的共价抑制剂。
编译:肖高铿
发布:2018-05-14
原文:Schulz, R., Atef, A., et al. (2018). Phenylthiomethyl Ketone-Based Fragments Show Selective and Irreversible Inhibition of Enteroviral 3C Proteases. Journal of Medicinal Chemistry, 61(3), 1218–1230. https://doi.org/10.1021/acs.jmedchem.7b01440
一. 前言
在畅销药里有一部分药物是通过共价键结合的药物,比如乙酰水杨酸、β-内酰胺抗生素、质子泵抑制剂(比如奥美拉唑)以及抗血小板聚集药物氯吡格雷。由于具有潜在副作用,药物设计时很少考虑使用含有共价结合基团的化合物1。但这种偏见最近有所变化,详见Singh(2011)的综述2《The resurgence of covalent drugs》。
共价结合当然有风险,但也带来不菲的收益,包括3-8:(1)延长与靶标的相互作用;(2)提高结合亲和力;(3)药代与药动的联动;(4)改善选择性。与基于片段的设计方法组合,共价结合可以用来9-12:(1)为抑制剂设计提供起始化合物;(2)探讨全新的蛋白结合位点;(3)提高发现蛋白配体的能力。采用共价结合策略使得结合位点很浅的蛋白也有可能找到合适的配体并进而发现苗头化合物、还可以进一步用质谱来验证这种结合。
不可逆的蛋白结合过程分两步:(1)首先形成可逆的初始非共价复合物EI;(2)初始的复合物EI进一步过渡为共价键结合的复合物E-I。抑制剂的药效用二级速率常数kincat/KI来表示。kincat反映了共价键的形成速率,而KI反映了通过非共价相互作用形成初始复合物EI的稳定性,整个过程可用下式描述:
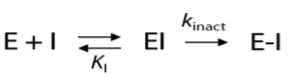
因此,共价药物设计的目标是:发现结合过程由KI而不是kincat驱动的共价结合片段。Schulz(2018)等人最近采用3D-药效团虚拟筛选成功实现这个虚拟筛选目标、发现选择性、不可逆的肠病毒3C蛋白酶(3C protease, 3CPro)共价结合抑制剂。据报道,3CPro与(1)无菌性脑膜炎;(2)心肌病;(3)1型糖尿病;(4)老年痴呆等疾病有关。3CPro一直以来是也肠病毒抑制剂设计很前途的靶标,但因为它的结合位点很浅,所以它的小分子抑制剂设计一直是个难点。目前针对该靶标活性最强的抑制剂是天然底物的肽拟物(Peptidomimetic analogues),它们通过Michael加成与催化位点的半胱氨酸共价结合使3CPro失活。然而,这些抑制剂的肽类性质使得其PK性质很差、限制了该类化合物的临床应用前景,因此进一步开发非肽类抑制剂是很有必要的。本案例的主要目的是与大家分享Schulz等人的研究过程与思路。
二. 方法与结果
2.1 共价药效团模型与片段筛选
Ligandscout的药效团模型通过引入了共价键这个药效团元素(covalent bond feature)来表征共价相互作用特征,见图1。
图1. Ligandscout的药效团元素
首先收集文献报道的共价结合的子结构,虽然文献很多,但这些共价结合配体或子结构的反应机理无非是以下之一:(1)Michael加成;(2)乙酰化;(3)形成缩醛或缩酮;(4)烷基化;(5)生成硼酸酯;(6)磺酰化。接着,将不适于理性设计的子结构刨除,包括:(1)反应性过强的片段,比如卤代烷没有结合专一性而被放弃;(2)需要转化后(比如酶反应)才能共价结合的片段;(3)需经复杂转化的子结构;(4)由于离去基团而具高度反应性的片段等。最后将这些精选的”弹头”用SMARTS编码内置于Ligandscout以供在虚拟筛选中进行模式匹配。共价键特征用一个黄色的球来表示(图1),位于配体与蛋白发生共价键的原子之间。
2.2 药效团模型的生成
用MOE 2014.09从PDB上下载蛋白结构3ZZ6、3ZZ7、3ZZ8、3ZZ9、3ZZA、3ZZB、3ZZC、3ZZD并进行结构准备。将准备过的蛋白结构读入到Ligandscout的Structure-based模块,自动生成药效团,并人工确认化学合理性;将这些药效团在Ligandscout的alignment模块里进行比较、识别它们的公共特征药效团(shared feature pharmacophore),结果见图2B。

图2. 3CPro共价药效团模型。(A)共价药效团特征及其匹配的共价结合子结构2D与3D图,从上到下分别是Michael受体,α-酮酰胺,&alpha-卤代甲基酮。(B)验证过的3D药效团模型,其中橘黄色球是共价相互作用、黄色球是疏水相互作用、红色箭头为氢键受体、绿色箭头为氢键供体。(C)活性化合物叠合到3CPro 3D药效团模型上。
2.3 药效团模型的验证
Reich et al、Webber et al.以及Johnson et al.报道了33个生物活性(kinact/KI大于等于100M-1S-1或KI 小于 1 μM)的非肽类3CPro抑制剂。以这33个已知活性化合物为基础,采用全新设计的方法生成1771个decoy分子。验证结果表明,药效团模型与33个活性化合物中的29个匹配,而仅与1771decoy分子中的20个匹配,这表明模型的具有良好的敏感性(0.87)与非常高的专一性(0.99), 如图3所示。
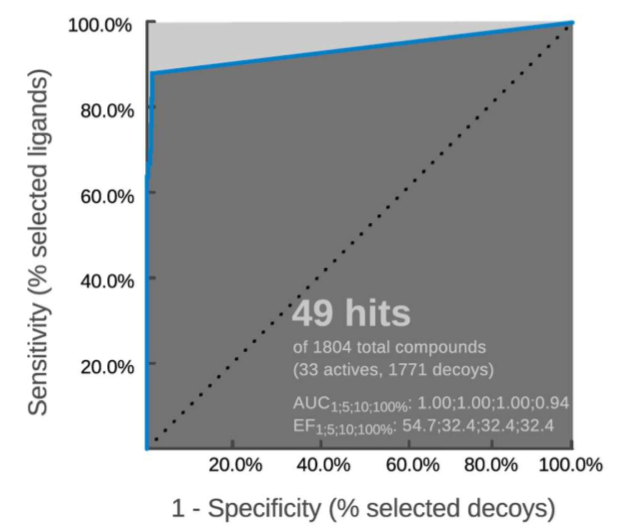
图3. 药效团验证之ROC曲线
总的来说,药效团模型的方法学验证说明该模型是可靠的,可以用于下一步的大规模虚拟筛选。更多的方法学验证说明,请参考我们的博文:《如何进行虚拟筛选的方法学验证》。
2.4 3D-药效团虚拟筛选
虚拟筛选的化合物库是内部自用的。在虚拟筛选之前先用Rule of 313对数据库进行过滤,以确保化合物具有片段的特征;再过滤掉其中的PAINS化合物14。最后获得的数据库含3000个片段样化合物,再用2.3验证的药效团模型进行3D药效团虚拟筛选,从中命中47个化合物。
2.5 共价对接模拟
用GOLD将47个化合物进行共价与非共价对接计算,其中有19个片段的初始构象与共价结合构象可以与3D药效团模型配体。
2.6 生物学验证
上一步19个片段包含了12种不同的化合物类型。每种化学类型各选取一个化合物进行生物活性测试(3CPro酶学测试)。其中6个片段在650µM水平对3C pro的抑制率达到30%以上。片段F1显示出了浓度依赖于时间依赖的活性特点,其IC50为355± 30 µM。然而,F1结构的化合物不稳定性限制了其进一步开发的价值,因此需对F1进行骨架跃迁以期望发现活性更好的化合物。
2.7 骨架跃迁与C5化合物的发现
对F1化合物进行基于结构的骨架跃迁,将F1与3CPro共价结合的酮片段、以及与S1口袋发生氢键相互作用的酰胺用SMARTS进行模式匹配,最后发现了五元杂环化合物(图4 A)。匹配的部分用蓝色表示。将这些化合物进一步与药效团进行匹配,其中有5个化合物(图4 B)有商品可供购买。这5个化合物可以可以从同一个供应商处购买、因此进行了采购、并进行生物活性测试。

图4. F1化合物的骨架跃迁发现了化合物C5
其中C5化合物在650µM水平可以完全抑制3CPro的活性,而其它化合物的抑制率均低于20%。然而C5并不含有显而易见的共价弹头,其结构也与其它化合物相似,尤其是C4与C6。高分辨质谱证明C5通过硫酚的离去而与3CPro发生共价加成, 见图5。
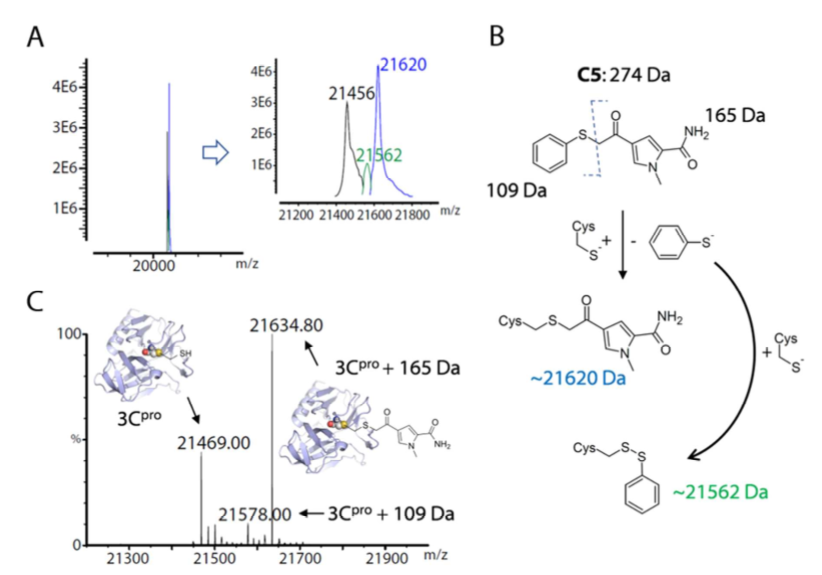
图5. C5通过硫酚的离去与蛋白发生共价加成
根据质谱数据,作者修订了C5的结合模式,如图6所示。C5以可逆的硫半缩酮与蛋白通过非共价相互作用形成起始的复合物结构,主要通过酰胺片段与S1口袋的His161、Thr142形成关键的氢键相互作用;而酮片段与催化位点Cys147非常接近(图6 A、B)。此外,在起始的复合物结构里半胱氨酸硫醇也可攻击C5酮部分的α-碳。硫酚被替换的SN2反应生成了在质谱中检测到的不可逆硫醚产物。共价对接表明硫醚产物的酰胺部分能够在S1口袋中形成关键的氢键,该保守的结合模式也证实了基于3D药效团的结合假说(图6C)的合理性。
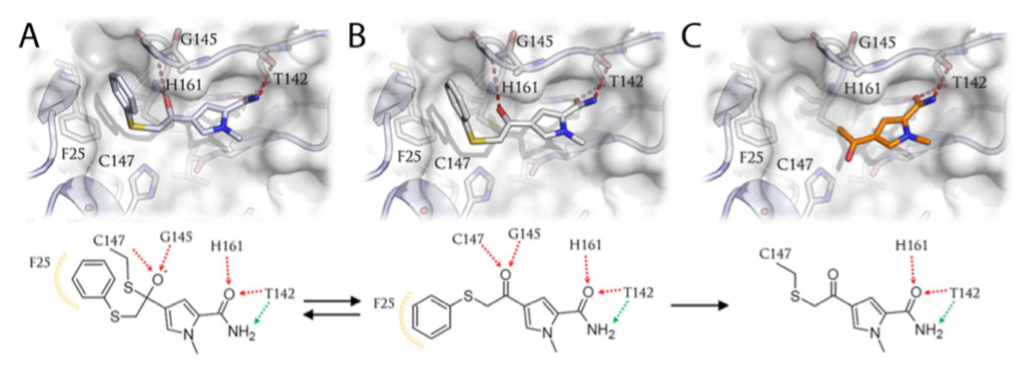
图6. C5与3C Pro的可能共价结合模式:复合物的3D(上)与2D(下)图示。相互作用通过药效团来展示:黄色球代表疏水相互作用、橘黄色代表共价键形成、绿色与红色箭头代表氢键供体与受体。
总的来说,生物活性以及由此发现的活性悬崖(注:由于篇幅所限,没有讲述活性悬崖)是由于C5通过α-苯硫基甲基片段形成不可逆的硫醚来实现,这是α-苯硫基甲基片段首次被发现有这种功能。至此,作者的标题结构类型化合物C5出现了。
2.8 片段的优化与弹头分析
为了进一步研究C5的结合机理以及提高化合物对3CPro的抑制活性,作者进一步进行了合成片段的优化工作。在提出的结合模式基础之上,让C5在3CPro活性位点里生长以占据更多的结合位点空间,其中包括图7 A所示的S2与S4口袋。C5的吡咯环N提供了合成的起始点,可以往上接不同基团去占据甲基所不能占据的空间。C5的这个衍生物设计采用基于结构的从头设计策略,旨在尽量让衍生物占据结合口袋而不改变原有的结合模式。从头设计产生了2000个N-烷基化的C5衍生物,采用药效团模型对化合物进行虚拟筛选,命中的化合物要求能与S2结合口袋Leu127发生疏水相互作用(图7 B)。3D药效团命中的化合物进一步用分子对接考察三种结合态,以确保化合物在S1结合位点里会形成关键的氢键相互作用(图7 C)。最后选择化合物其侧链是有商品可供购买的卤代烷基砌块,如图6D所示。
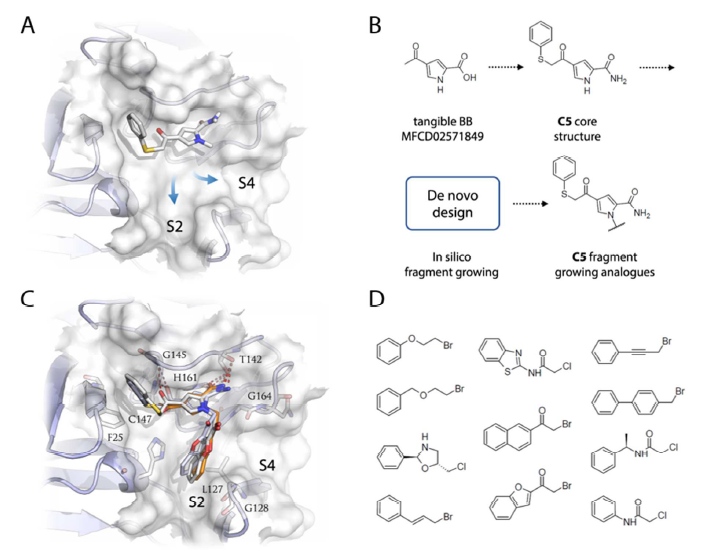
图7.采用从头设计策略让C5在3CPro结合位点里进行片段生长。(A)C5的结合构象以及在结合位点里的S2、S4生长取向;(B)C5片段生长策略从一个合成前体出发,采用基于结构的从头设计策略进行片段生长获得C5衍生物数据库,在吡咯环上具有不同的取代基;(C)分子对接表明,C5片段生长的产物占据了S2结合位点。呈现了三种结合状态:初始复合物(白色棒状),硫半缩酮(浅蓝色棒状)与最终的硫醚(黄色),红色虚线是氢键相互作用;(D)从头设计用到的商品可供购买的砌块(building block),它们填充到了S2口袋。
最后,作者设计了图8合成路线,并合成了化合物。在后续的研究中,作者探讨的7a与C5的结合模式差异等诸多内容,因为与本文的药效团虚拟筛选关系不大,这里就不再叙述。
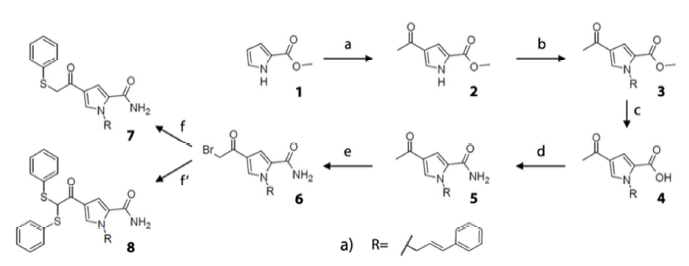
图8.C5衍生物的合成
三. 总结
- 生成含有共价键相互作用特征的基于结构的药效团模型
为了发现KI-驱动、专一性的3CPro共价抑制剂, 作者采用基于结构的策略开发了包含共价特征的3D药效团模型。
- 药效团模型的方法学验证
用活性化合物与decoy化合物对药效团模型进行验证,确认模型具有好的敏感性与非常好专一性。
- 药效团虚拟筛选
用验证过的模型对包含3000个化合物的数据库进行了虚拟筛选,命中47个化合物。
- 分子对接研究
47个化合物进一步用分子对接确认其起始结合模式与共价结合模式与药效团模型是一致,19个化合物满足这个条件进入下一步测试。
- 生物学验证发现F1结构
19个化合物属于12种不同的结构类型,每个类型取一个化合物进行测试,发现F1结构。
- 骨架跃迁与C5化合物的发现
F1结构化学不稳定,需要进行骨架跃迁以发现结构稳定但保留活性的化合物,结果发现了标题的结构类型C5化合物。
- 从头设计与结构优化
采用基于结构的从头设计策略设计C5的衍生物,再用药效团进行虚拟筛选、分子对接确认结合模式,合成一系列化合物探讨构效关系、结合模式分析…
为了发现KI-驱动、专一性的3CPro共价抑制剂, 作者采用基于结构的策略开发了包含共价特征的3D药效团模型。
用活性化合物与decoy化合物对药效团模型进行验证,确认模型具有好的敏感性与非常好专一性。
用验证过的模型对包含3000个化合物的数据库进行了虚拟筛选,命中47个化合物。
47个化合物进一步用分子对接确认其起始结合模式与共价结合模式与药效团模型是一致,19个化合物满足这个条件进入下一步测试。
19个化合物属于12种不同的结构类型,每个类型取一个化合物进行测试,发现F1结构。
F1结构化学不稳定,需要进行骨架跃迁以发现结构稳定但保留活性的化合物,结果发现了标题的结构类型C5化合物。
采用基于结构的从头设计策略设计C5的衍生物,再用药效团进行虚拟筛选、分子对接确认结合模式,合成一系列化合物探讨构效关系、结合模式分析…
总的来说,本文涉及的靶标3CPro结合位点很浅,不适合开发非共价结合的有机小分子抑制剂,目前仅有肽类共价抑制剂。作者通过共价虚拟筛选、实验验证、结构优化等手段最后发现不可逆抑制剂,为此类靶标的共价抑制剂发现提供了一个新的虚拟筛选实践案例。在本案中,虚拟筛选为高效化学与生物学工作提供了保证,最终发现苗头化合物或先导物。
四. 文献
- Nakayama S, Atsumi R, Takakusa H, et al. A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab Dispos. 2009;37:1970.
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307.
- Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20:1061.
- Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discov. 2010;5:305.
- Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2008;47:2003.
- Smith AJ, Zhang X, Leach AG, Houk KN. Beyond picomolar affinities: quantitative aspects of noncovalent and covalent binding of drugs to proteins. J Med Chem. 2009;52:225.
- Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Futur Med Chem. 2010;2:949.
- Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J Am Chem Soc. 2013;135:5298.
- Erlanson DA, Braisted AC, Raphael DR, et al. Site-directed ligand discovery. Proc Natl Acad Sci U S A. 2000;97:9367.
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548.
- Becker D, Kaczmarska Z, Arkona C, et al. Irreversible inhibitors of the 3C protease of coxsackie virus through templated assembly of protein-binding fragments. Nat Commun. 2016;7:12761.
- Cardoso R, Love R, Nilsson CL, et al. Identification of Cys255 in HIF-1 alpha as a novel site for development of covalent inhibitors of HIF-1 alpha/ARNT PasB domain protein-protein interaction. Protein Sci. 2012;21:1885.
- Jhoti H, Williams G, Rees DC, Murray CW. The ’rule of three’ for fragment-based drug discovery: where are we now? Nat Rev Drug Discov. 2013;12:644.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719.
五. 相关主题
- Ligandscout论文集
- Ligandscout教程 | 共价键药效团模型与虚拟筛选
用Ligandscout发表的文章已经有1000多篇,我们为您精选了一部分供您参考:《Ligandscout论文集》
以EGFR Kinase-Afatinia复合物为例来演示如何用Ligandscout基于结构的方法生成带共价键特征的药效团模型、以及如何用该药效团模型进行虚拟筛选。点此阅读原文。


