
摘要:2011年,阿斯利康开始对其研发战略进行重大调整,旨在提高研发生产率,该水平低于2005-2010年的行业平均水平。修订战略的基础是将决策聚焦于五个技术决定因素(正确的靶标、正确的组织、正确的安全性、正确的患者和正确的商业潜力)。在本文中,我们分享了使用"5R框架"所取得的进展,希望我们的经验能够对其他公司解决研发生产率问题有所帮助。我们专注于靶标验证、苗头化合物和先导化合物的优化、药代/药动学建模和药物安全性测试方法的优化,这些方法有助于提高提名候选提名的质量,以及正确文化的发展,更为严格的、定量的决策鼓励我们"寻求真理"。我们还讨论了该方法的失败之处和经验教训。总体而言,5R框架的持续演变和应用开始对研发生产率产生影响,从候选药物的提名到III期临床完成的成功率从2005-2010年的4%提高到2012-2016年的19%。
编译:肖高铿
原文:Morgan, P. et al. (2018). Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nature Reviews Drug Discovery, 17(3):167–181. http://doi.org/10.1038/nrd.2017.244
大量文献证实制药工业的研究和开发(R&D)生产率一直在下降,有人提出了各种各样的原因。其中包括过度依赖数量而非质量而导致的”暴力”研发方法(例如,制造”容易”的化合物,而不是制造正确的化合物),狭隘的临床试验,日益挑战的监管障碍,以及在无意中对追求进度的行为比追求真理的行为给予更高奖励的文化。对此,有人提出了一些改进建议。Kola和Landis用2004年行业损耗数据进行的开创性分析强调了研发损耗的最常见原因是缺乏有效性和/或安全性,并建议在临床上更多地关注降低毒性风险、改进临床前模型以及展示足够的机理验证( proof of mechanism, POM )和概念验证(proof of concept)。 2010年,Paul等人进行了另一项基于行业损耗数据的分析,该分析建议通过将损耗转移到较早的阶段来减少循环时间和成本。最近,Scannell等人提出了预测的有效性和定义明确的临床前模型对于期望的疾病设置的重要性——这两个因素可能产生更大的成功。
2011年,阿斯利康开始对其研发战略进行重大修订,旨在提高其研发生产率,其研发生产率低于2005-2010年的行业平均水平;从候选药物提名到临床III期完成的成功率为4%,而根据药物研究中心(CMR)国际中心的数据,该行业的平均成功率为5%。分阶段来看,该公司总体上与CMR行业平均水平一致,但II期的成功率特别低,为15%,而行业平均水平为22%。在本文发表之前已有文献深入讨论过上述策略,并产生了”5R框架”(见Figure 1)的制定和实施。5R框架将决策聚焦于五个技术决定因素:正确的靶标(right target)、正确的组织(right tissue)、正确的安全性(right safety)、正确的患者(right patient)和正确的商业潜力(right commercial potential)。该框架与上面讨论的一些R&D生产率分析一致。现在,阿斯利康的科学家们所涉猎的疾病领域比以往更少,但是专注于对疾病的生物学和机制更加深入地科学理解。公司还对其靶标选择和验证、先导化合物生成、药代动力学/药效学(PK/PD)建模、患者的分类(Stratification)和生物标志物的能力进行了大量投资。本文比较了2012-2016年和2005-2010年公司业务组合的特征数据,以及2012-2016年期间项目失败的教训,讨论了这些变化是如何提高生产率的,这些教训也突出了有待进一步改善的领域。与最初的5R框架文件一样,所分析的项目包括2012-2016年期间在阿斯利康IMED生物技术单元和后期发展的组织机构中做出的里程碑式决定的所有项目(92个小分子和10个大分子)。

Figure 1. 5R框架. 总结了5维框架的关键特征:right target, right tissue, right safety, right patent and right commercial potential.
理解正确的靶标
5R框架向科学家们提出挑战,要求他们不仅在项目开始时,而且在整个项目过程中验证或否定他们的科学假设。对靶标基础理论更强烈的、更多的关注导致了在发现阶段组合投资中项目数量的显著减少(Figure 2a)。在2005年到2010年间(在5R框架启动之前),启动了287个小分子发现项目,在2012年到2016年间减少到76个项目。此外,备份项目的数量从业务组合的28%减少到不到7%。
聚焦于对生物学的理解驱动的靶标选择还导致了管线靶标类别组成的改变(Figure 2b)。激酶已经成为主要的靶标类别(从21%增加到36%),这是由于对肿瘤学的重视所致,此外靶向G蛋白偶联受体的项目比例从25%下降到5%。以离子通道为靶标的项目比例也有所下降,从8%降至2%。其它类别的靶标数量有所增长(从13%提高到21%)。其它类别靶标包括蛋白-蛋白相互作用、转运蛋白、生长因子、转录因子、表观遗传酶、磷酸酶、解旋酶和去泛素酶等等,通常人们认为这类靶标难以发现对应的小分子药物。
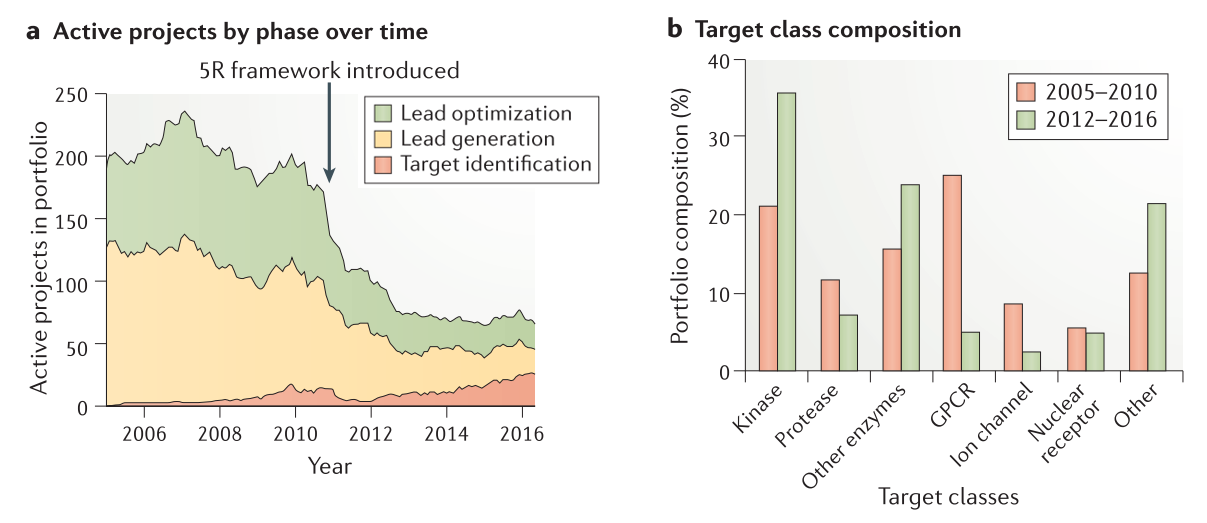
Figure 2. Number and phase of active preclinical projects in the portfolio from 2005 to 2016 and their target class composition
除了靶标类别组成的改变之外,越来越多的项目寻求具有新作用机制的化合物。例如,许多激酶和酶的项目力图确认变构调节剂而不是经典的竞争性抑制剂。这种靶标多样性的增加要求先导化合物发现策略的演进。
为了支持对 “正确靶标”的理解我们已经投资了几个平台,包括干细胞生物学、表型筛选、精确的基因组编辑和基因组学。通过建立来自外部来源以及我们自有临床研究的可搜索、获取的人组织生物样本库,改善了对人组织和人源诱导多能干细胞(iPSC)的获取可以改善生物的人源化(biological translation )。因此,复杂的表型筛选现在占了新项目的25%。生物测定日益增长的复杂性要求更多地采用诸如化学生物学、化学蛋白质组学、化学注释的筛选平台和细胞热转移测试技术来鉴定、筛选化合物。尽管我们已经提高了利用这些技术进行基于细胞的表型筛选的能力,但这些筛选的方法的转化仍然是关键的瓶颈和主要的资源支出。
我们和其他公司都在使用CRISPR和转录激活子样效应核酸酶(TALEN)技术进行基因组编辑,以创建更相关的细胞和动物疾病模型。CRISPR试剂库也可用于选择性抑制、激活或删除任何用于筛选的基因,并用来证明靶标和通路的有效或是无效。
MTH1是一个备受关注和研究的肿瘤靶点 ,最近MTH1(也称为NUDT1)的研究也受到基因组编辑技术的影响。在这些研究中,癌细胞在MTH1完全沉默后保持存活,表明先前公开的MTH1抑制剂不是通过MTH1抑制而是通过其它机制来杀死细胞。这些数据让我们快速的做出停止MTH1抑制剂项目的决定,其它团队也证实了此事。在第二个实例中,我们应用基因组编辑来产生盐诱导激酶(salt-inducible kinase, SIK)的三种亚型激酶失活突变体,以鉴定负责介导炎症反应的亚型。这些数据使项目能够快速地将化学集中在正确的酶亚型上,并建立适当的选择性筛选方法。这些例子强调了基因组编辑是如何比以往更早地建立对靶标的信心或证明靶标无效而放弃一个靶标。
展望未来,下一代测序技术的进步促使人们对基因组学进行了大量的投资,以帮助在大量患者群体中识别新的、经过基因验证的靶标。包括我们和葛兰素史克(GlaxoSmithKline)在内的几家公司指出,选择经过基因验证的药物靶点可以提高临床开发的成功率。例如,Regeneron Pharmaceuticals与Geisinger Health System(美国综合健康提供者)合作对60,000名患者的外显子测序表明血管生成素相关蛋白3(ANGPTL3)失活在预防心血管疾病中的潜在作用。目前已有大量公共和私人力量积极地对大量患者和健康受试者的基因组进行测序,包括英国的10万个基因组项目、美国国立卫生研究院资助的百万基因组精准医措施(见进一步信息),以及我们自己也投入资源分析200万个患者样本基因组。来自这些方法的数据,加上基因编辑技术,应该可以进一步改进靶标验证、靶标选择、疾病理解和药物发现生产率。
为“正确的靶标”发现先导化合物
随着对靶标验证的关注增加,鼓励团队使用多种技术来最大化找到可以转换为先导化合物的苗头化合物的概率。化学工作,尤其是高通量筛选(HTS),仍然是新项目的先导化合物产生的主要策略。随着投资组合规模的缩小,HTS的数量已从约2010年前的每年40次左右减少到现在的每年20次左右。然而,筛选的复杂性一直在增加,通过使用诸如高内涵成像、高通量电生理学、高通量荧光激活细胞分选、无标记检测、转录组学和声学质谱等技术的使用来测试化合物活性的多个参数。
我们持续扩大化合物收集的规模和范围,已经添加了600,000多种化合物。重点在于添加可能对新靶标有活性的化学类型,包括添加定向的子集比如片段、sp3样化合物、天然产物样分子、大环、核苷和共价修饰剂。通过与同行公司的化合物共享或交换,也有可能获得新的化学多样性—鉴于化合物库从来一直被视为高度专有,一些人可能认为这是异想天开。例如,我们与Bayer的化合物库共享合作计划,允许一个公司访问另一个公司的全部化合物用于HTS。迄今为止,与该联盟合作的项目中有75%的项目发现了从其他公司的化合物库中未发现的先导化合物。我们还进行了一系列的化合物交换,其中最大的一次是与赛诺菲交换了21万个化合物。最后,我们作为向欧洲联合化合物库捐赠化合物的七家公司之一,使得每个合作伙伴能够获得500,000种新化合物。这些伙伴关系以相当低的成本增加了化学库的多样性,也使得我们可获得的高质量分子数量超过4百多万个。
除了HTS之外,和其他人一样,我们还集成了其它的筛选方法,包括DNA编码库筛选、基于片段的先导化合物产生、基于结构的药物设计和虚拟筛选(例如Table S1)。重要的是,团队现在平行使用多个筛选方法,而不是多个方法串联筛选,以获得最广泛的化学洞察力以便产生先导化合物。慢性炎性疼痛的靶标蛋白酶激活受体2(protease-activated receptor 2 , PAR2)的第一个有效的变构拮抗剂的识别就是一个好例子。通过与Heptares Therapeutics合作,我们制备了热稳定的受体,并通过基于生物物理的片段筛选策略,我们识别出了结合于变构位点有效的、选择性的配体(见Table S1,10)。通过使用DNA编码库筛选(Table S1,4)平行地发现了与备选变构位点结合的另一系列苗头化合物。
在引入5R框架之前,只有23%的新项目交付了质量足以支持进行先导化合物优化的先导化合物。修订后的hit-to-lead策略将这一比例提高了一倍多,达到48%,转化为业务组合模型,根据该模型,仅需要两个先导化合物发现项目就可以孵化出一个先导化合物优化项目,而不是之前所需的四个项目(Figure 3a)。从项目开始到先导化合物优化的循环时间从过去的26个月减少到现在的19个月(Figure 3b)可能是由于我们减少了项目并使用并行的先导化合物发现方法。
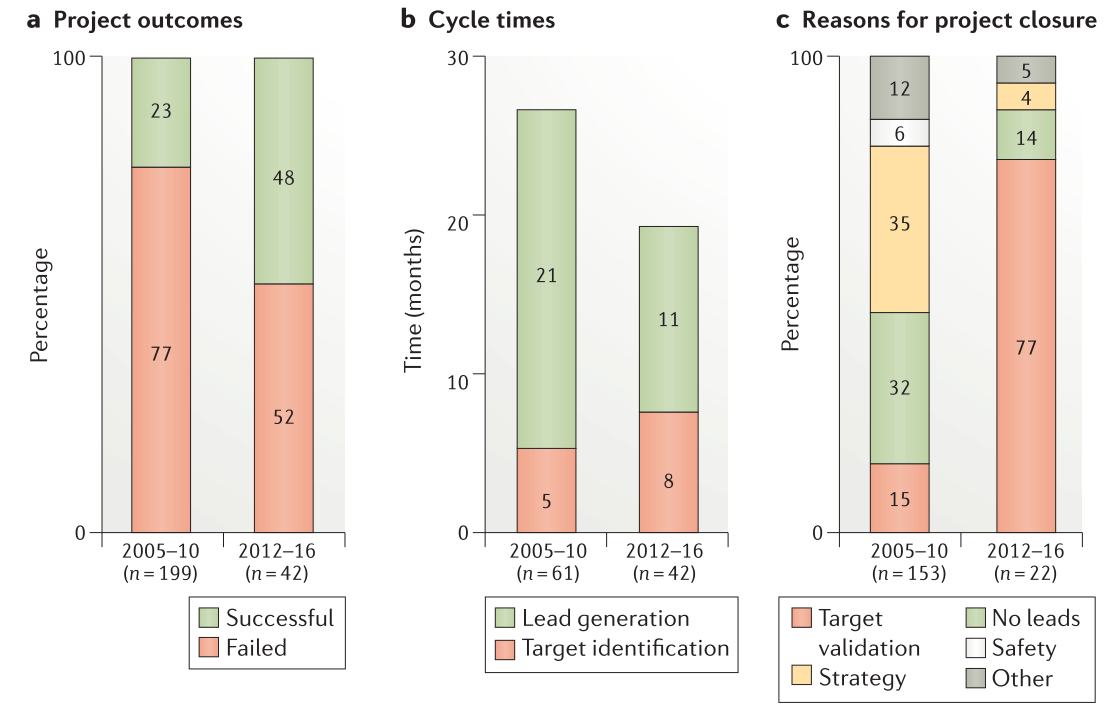
Figure 3. Metrics for preclinical projects reaching the lead optimization phase.
将2012-2016年与2005-2010年的队列进行比较,从项目启动到先导化合物优化完成的早期药物发现项目的失败原因也发生了变化(Figure 3c)。毒性试验的早期使用(也参见下文”正确的安全性”一节)使得由于分子安全性导致的项目关闭数量减少,在项目进行期间持续关注具有相同母核结构的疾病领域,可以将由于策略变化导致的项目关闭减少到5%以下(Figure 3c)。对先导化合物识别的改进使得由于没有发现先导化合物而导致的项目关闭从32%减少到14%(Figure 3c)。虽然如此,但这仍然是持续改进的重要领域,特别是具有挑战性但获得高度验证的靶标时。例如,GTPase KRAS长期以来一直是重要但具有挑战性的肿瘤靶点,尽管已经筛选出苗头化合物,但是这些苗头化合物扩展到高质量的先导化合物系列和临床候选化合物依旧困难,如同Cox及其同事在最近的综述中描述的一样。令人鼓舞的是,最近有几篇文献描述了G12C突变蛋白的共价修饰物,可能使靶向KRAS的新小分子方法的前景更加光明。有趣的是,新的模式,如反义寡核苷酸和抗KRAS疫苗也正在探索作为替代方法以达到这一重要的目标。今后,具有挑战性但经过验证的靶标仍将是优先事项,我们预计将持续投资于新的小分子筛选策略和其他治疗方式(如寡核苷酸和蛋白水解靶向嵌合体PROTAC)作为找到这些重要靶标的解决方法。
靶标验证是目前早期发现项目终止的主要原因,从15%上升到77%(Figure 3c)。这是由于人们越来越多的关注于实验产生证据的结果,这些证据可以建立对科学假说的信心或用于证明假说无效。尽管我们预计靶标验证将仍然是项目终止的主要原因,但是使用诸如基因组编辑和基因组学之类的能力以及改进人源化的临床前模型将有望减少在该领域的损耗。选择正确的靶标仍然是5R中最重要的,也是我们在药物发现方面做出的最重要的投资决策。如果所调控的靶标不是”正确的靶标”,那么选择性的高质量分子将永远不会成为药物,当然,只有当作于用于该靶标的药物被成功批准时,靶标才会被真正地验证。
理解正确的组织
先前确认可以提高项目成功率的一个关键参数是靶标结合证据(Target engagement evidence)或POM证据,定义为候选药物在人体中以预定的、定量的水平与靶标相互作用,并表现出功能效果。有助于PK、PK/PD、吸收、分布、代谢和排泄等ADME性质理解的数据不仅对改善先导化合物和候选药物的选择质量是至关重要的,而且还有助于设计最适当的临床研究以证明靶标结合(target engagement)。 ADME和PK性质的改善已经从2000年的HTS阶段推进到更加综合、基于模型的方法上,这些方法将ADME数据整合到人的PK/PD与有效剂量的预测里。人们已经开始将药物代谢和PK性质与分子设计相结,使得在选择候选药物时更加关注定量PK的弹性(scaling)、不仅可以预测、而且还可以与人的PK/PD模型和剂量预测相结合。
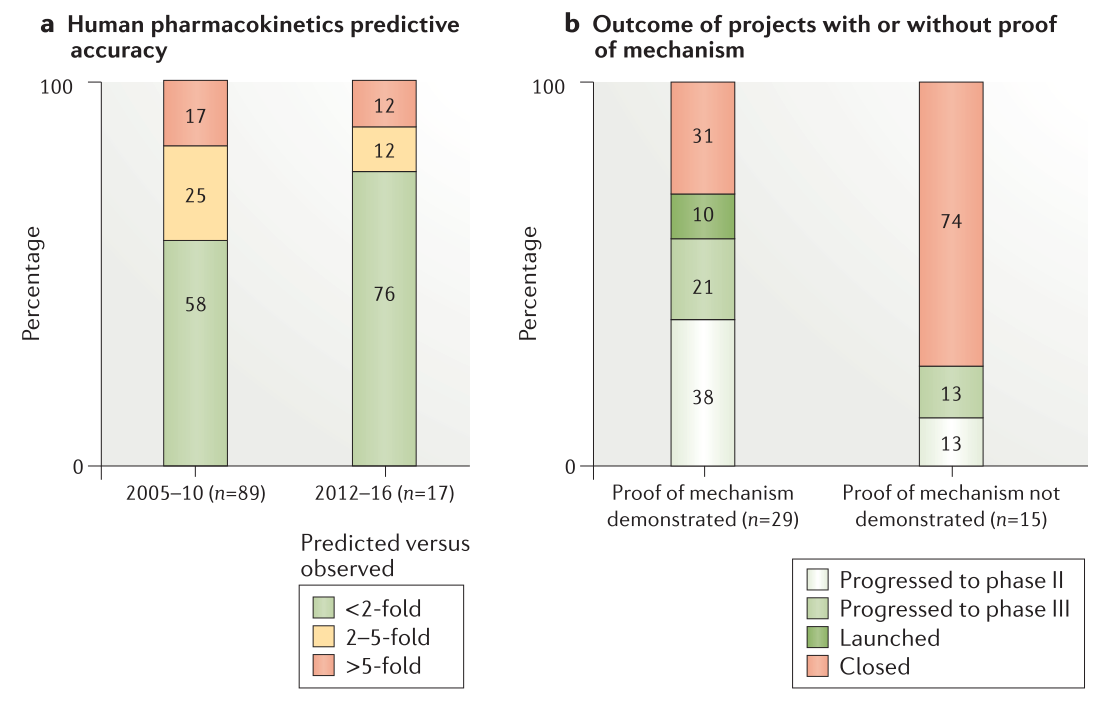
Figure 4. Human pharmacokinetics prediction accuracy and impact of proof of mechanism on project success rate
近期项目(2012-2016年)和历史项目(2005-2010年)的比较显示,PK预测值在PK实验值相差在两倍以内的化合物数量显著提升(76%对58%,Figure. 4a),这说明将ADME数据与人的PK/PD和有效剂量预测相结合的基于模型的方法的重要性。在预测上的改善与同行公司相当。预测的作用体现在临床执行上:其中PK或PK/PD预测不良的项目需要改变给药剂量或给药策略,甚至关闭项目。重要的是,对PK预测准确性的滚动评估表明,85%PK或PK/PD预测不佳的项目,项目团队在在其临床计划中不再调整或项目被延迟。
继续理解PK预测差的原因仍然是重要的,预测值和观测值之间存在五倍多的差异。在2012-2016年间的两个项目说明了仍需改进的领域。一个项目是AZD9496,一种小分子雌激素受体阻断剂),它的人体半衰期(1-2小时)比预期的(6小时)短,这是由于对清除率的低估。进一步的分析表明,高度可变的蛋白结合率导致清除预测的范围过宽,被低的分布体积进一步放大了预测误差。改进的定量体外清除率和蛋白结合率预测模型有助于开发更灵敏、全面的PK模型。第二个项目是AZD1208,一种丝氨酸/苏氨酸蛋白激酶PIM的小分子抑制剂,预测具有P4503A(CYP3A)诱导机制因而PK性质差。临床前研究表明,CYP3A4介导的代谢在人体中占主导地位。在临床前使用的标准CYP诱导试验中,在HepaRG细胞或人肝细胞中没有表现出任何的诱导潜力。在临床前PK和单剂量人PK研究中,该化合物显示出与剂量成比例的暴露,半衰期长达>30小时。然而,在多剂量人体研究中,暴露随着时间和剂量而减少,表现出时间依赖性的PK特征。初步分析表明这种CYP诱导机制很独特,不同于传统的诱导机制,正在评估几种替代方法以进一步理解该机制。重要的是,该机制在目前使用的体外系统中并不存在,因此这些模型不能鉴定具有类似诱导机制的其它物质。
在改进PK预测的同时,关注”正确的组织”意味着团队需要理解PK/PD以及靶标处的药物暴露与随后的下游生物效应之间的因果关系。PD生物标志物可以更好地界定在预期耐受剂量下与靶标结合的程度和持续时间。PD生物标志物使用的主要目的是根据临床POM做出进行或不进行(go/no go)的决策,确定分子能否充分地与靶标结合。此外,阳性POM表明分子具有足够的PK暴露和安全边界在良好耐受剂量下与靶标结合。2012-2016年期间,我们有29个POM正值,而在2005-2010年间的POM值不到5。POM阳性的项目比起没有展示出POM的项目有更高的比例进入到II期(38%)、III期(21%)或上市(10%)阶段(Figure 4b)。在POM阴性项目进展到II期(13%)和III期(13%)的项目中,四个项目中的三个(AZD8871、AZD2115和PT010)在原理验证研究中使用相对简单的疗效终点:1秒内用力呼气量的改善。研究人员认为这比测量肺中的靶标结合更加容易,且足以确定项目的进行/不进行决定标准。 4个项目未尝试评价POM (AZD8931、MEDI8968、AZD9773和AZD4017)。其中三个项目在执行5R框架之前已经进行了概念验证研究,最后四个项目均因疗效不佳而失败(Table 1)。
突出POM决策重要性的实例可通过比较经由TLR7或TLR9靶向Toll样受体(TLR)通路的两种吸入药物来看出。AZD8848是一种TLR7激动剂,AZD1419是一种TLR9激动剂。在临床研究中评估作为干扰素应答指标的生物标志物CXC-趋化因子配体10(CXCL10)用来评估TLR的靶标结合。30μg剂量的AZD8848导致CXCL10水平增加2.5倍,并且抑制了50-60%对过敏原的过敏反应,证明了充分的靶标结合和阳性POM。然而,在该剂量下,患者也经历中度或严重流感样症状,导致疗效与不良事件没有剂量分离(S2 Figure)。相比之下,AZD1419在痰液和血浆中CXCL10水平增加了>2.5倍,但只有在试验的最高剂量下才观察到与剂量相关的流感样症状。这可以证明肺中的靶标结合与全身不良事件剂量之间显著地分离(S2 Figure, part b)。这使得能够在达到POM的剂量下区分两种化合物,并导致AZD8848项目的终止和AZD1419的继续开发(Table 1)。
定量PK和PK/PD建模的总体改进有助于提高候选药物的质量、提升对PK/PD理解的更大信心,以及提高对靶标结合的研究能力。5R框架该部分应用的三个明确的表现是:(1)更高的人体PK预测精度(Figure 4a),(2)增加的临床POM (Figure 4b, Table 1)和(3)减少由于PK/PD问题而导致的失败(Figure 5b)。
理解正确的安全性
通过引入”正确安全性”作为5R框架的核心要素,安全性评估的变化有助于识别早期的临床前安全性信号,并整合体外和体内数据,用于将来人体使用的定量风险评估。例如,我们现在通过探索靶标在健康和疾病环境中的生理作用来考虑靶标相关的安全责任,从而导致了对靶标相关安全风险的早期假设检验。对先导化合物生成阶段分子进行计算预测和体外安全性试验评价的目的是:了解其对关键器官的潜在作用。使用人源系统的新型模型,比如3D肝微组织或人iPSC-衍生的心肌细胞,可以早期集成到项目中以降低肝或心脏毒性风险,而这正是2005-2010年间由于安全性原因导致临床失败的两个主要原因(Figure 3c)。
随着项目越来越接近候选化合物选择阶段,需要进行跨物种的体外和体内测试用于指导决策。在物种特异性效应、预测的治疗范围和可能的临床剂量方案的范围内,对安全风险是否可接受进行评估,包括广泛的建模和模拟。例如,使用体外肠器官的跨物种比较使得溴域蛋白4(BRD4)抑制剂AZD5153在进一步开发前被终止。在亚治疗剂量的狗中观察到严重的胃肠道毒性,导致肠上皮萎缩和治疗边界的侵蚀。为了进一步探讨这一点,使用肠干细胞微组织的3D器官来确定大鼠、狗和人肠细胞对一组BRD4抑制剂的相对敏感性。发现在所测试的所有物种中,人体器官是最不敏感的(S3 Figure)。这些数据支持OTX015的临床转化,OTX015是一种竞争性BRD4抑制剂,它在大鼠中显示毒性但在人器官中不显示毒性,并且重要的是其在暴露于比大鼠最大耐受剂量高四倍的剂量时对人耐受,没有显示出剂量限制的胃肠毒性。
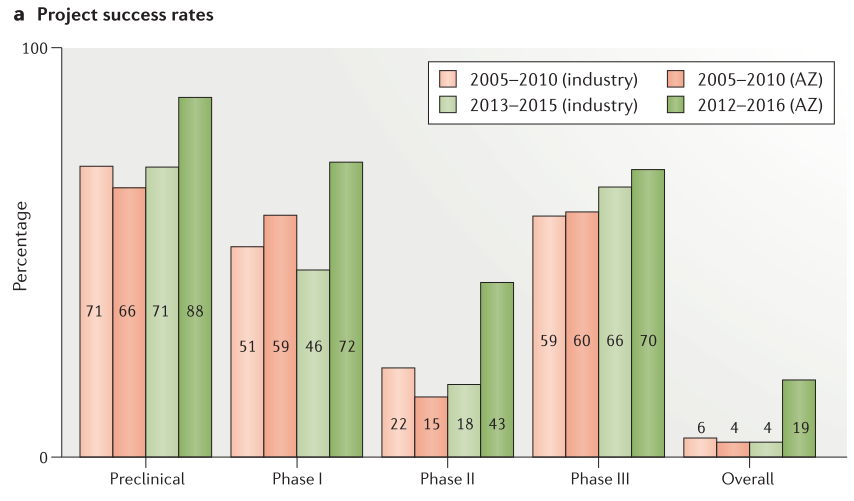
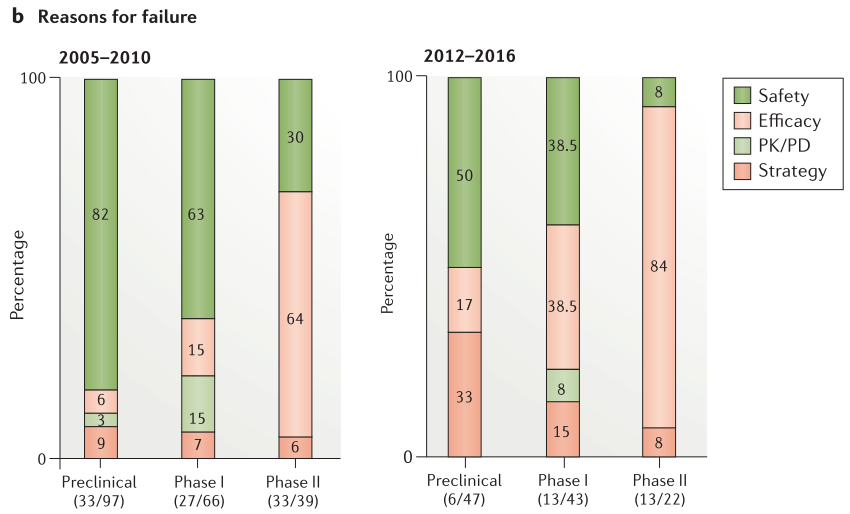
Figure 5. Project success rates and reasons for failure
候选药物提名后的临床前成功率有所提高,从2012年至2016年的项目成功率为88%,而2005年至2010年的项目成功率为66%(Figure 5a)。 在2012年至2016年期间,47个项目中只有6个在临床前中失败,这6个项目中有3个由于安全原因而失败。与之相比,在2005年至2010年期间,82%的临床前项目由于安全性问题而失败(33个项目中有27个)。在临床阶段与安全性相关的失败数量和百分也大幅减少:I期(5个项目,38%的关闭),II期(1个项目,8%的关闭),并且通常由于意外的、特殊的安全原因而发生(Figure 5b)。
观察当前安全性失败的原因,可以发现我们对某些领域的理解仍然很差。例如,在临床前开发过程中终止的一个分子在遥测研究(Telemetry study)期间显示心电图R波部分的振幅呈剂量依赖性降低。随后的研究表明,这是一个以前没有发现的与靶标相关的作用,可能在治疗剂量下会发生。鉴于对这一罕见的心血管发现如何转化为临床环境缺乏信心,该项目被终止。这说明了心脏功能的一个领域,R波振幅,在人体相关模型中定义不清。在临床上,单次给药黑色素浓缩激素受体1(melanin-concentrating hormone receptor 1)拮抗剂AZD1979后,几名受试者的丙氨酸转氨酶(ALT)迅速升高。在任何临床前毒理学研究中都没有观察到丙氨酸氨基转移酶升高,也不能归因于任何特定的机制,从而导致项目终止。继续完善我们的临床前肝毒性模型,以便更好地了解如何避免在未来类似的发现。另一分子是毒蕈碱受体拮抗剂AZD8683,在慢性阻塞性肺病(COPD)的临床IIa研究中,显示两名患者的肺功能意外但显著地降低。在任何临床前研究中均未观察到对肺功能的有害影响,对这一发现缺乏了解导致该分子终止研发。
尽管我们已经看到整个管道安全性相关的损耗有了很大的改善,但仍有一些特殊的或无法解释的毒性。我们和其他公司继续探索新的体外和体内模型,以提高临床的转化率。一个快速发展的领域是微生理系统(MPS),它使人源性细胞能够在生物工程芯片上培养,从而更紧密地复制组织微环境、循环流动和细胞-细胞相互作用。令人感兴趣的是MPS能否提高安全性临床转化,取代现有的体外模型,甚至可能减少或取代动物的某些安全性实验。
正确的患者
新兴科学不断提高我们在多个疾病领域发展与疾病亚组相关的生物标记物的能力。基于药物靶点表达或修饰的单基因生物标志物通常具有足够的预测性,可用于早期临床开发。例如,使用免疫组织化学检查程序性细胞死亡1配体1(PDL1)的表达已被用作识别癌症患者的一种方法,这些患者可能对针对免疫检查点蛋白程序性细胞死亡1(PDL1)或其结合伙伴PDL1的单克隆抗体(mABs)更为敏感。诊断试验的出现有助于理解PDL1的表达,其中三种最常用的方法在检测肿瘤细胞的表达时具有分析上的可比性。PDL1状态的临床效用已经得到证实,特别是在非小细胞肺癌(NSCLC)的一线治疗中,使用美国食品和药物管理局(US Food and Drug Administration )批准的伴随诊断测试时肿瘤细胞表现出50%以上PDL1表达的患者使用Pembrolizumab单药疗法比化疗具有临床效益。在晚期NSCLC治疗中,结合化疗或其他治疗标准无效的适应症,PDL1状态的临床价值不太明确。在这种情况下,补充诊断(如尿路上皮癌的二线治疗中的Ventana SP263)用于指导逐个病例的治疗决策,而不是将治疗限制在特定的患者亚组。
对致癌驱动突变作用的新认识也使得单基因突变作为选择性生物标记物在临床试验中得到广泛应用。例如,AKT1和PIK3CA的突变可以选择最有可能对AKT抑制剂产生反应的患者,如AZD5363(REF. 93)和MK 2206(REF. 94),以及磷酸肌醇3-激酶(PI3K)抑制剂,如Genentech的GDC0941或Novartis的BYL719(REF. 95)。基因拷贝数或基因扩增也被用来选择患者进行治疗。编码受体酪氨酸激酶的Met原癌基因的表达已被广泛用于识别在包括乳头状肾细胞癌和NSCL96-98在内的适应症中更可能对诸如Onartuzumab、Savolitinib和Capmatinib等Met抑制剂产生反应的患者。然而,这些研究并不总是成功的;Onartuzumab未能提高MET高表达或基因拷贝数的NSCLC患者的无进展生存率或总生存率。
在呼吸系统疾病中,为患者选择的生物标志物现在包括循环生物标志物,如免疫细胞类型。血嗜酸性粒细胞计数已被提出作为减少病情恶化的预测指标,并已成功用于识别最有可能对以白细胞介素-5(IL-5)通路为靶点的单克隆抗体治疗作出反应的严重哮喘患者,支持批准IL-5特异性单克隆抗体(mpolizumab)和重组单克隆抗体(reslizumab)用于有嗜酸细胞表型的哮喘患者。在基线血嗜酸性粒细胞计数较高的哮喘患者中,IL-5受体特异性单克隆抗体benlizumab在临床III期试验中也显示了哮喘年加重率的显著降低。然而,对于哮喘患者的IL-13通路,使用生物标记物选择应答患者的可靠性较差。在早期的试验中,在中到重度哮喘104名患者中,无论是潜望素计数还是嗜酸性粒细胞计数都能成功预测对IL-13特异性单克隆抗体-利布里基祖马b的反应,但当更多的患者接受试验时,这一结果并未得到一致的复制。
在初级护理环境中,通常不可能在治疗开始前将患者样本送到实验室进行检测。随着新的靶向治疗的发展,初级保健医师将需要快速的诊断测试,如点式护理设备,以根据患者的个人情况将合适的患者与最佳治疗方案相匹配。目前正在开发的例子包括结核病的分子遗传学检测、凝血功能障碍性出血的凝血酶原时间、痛风的尿酸水平以及呼吸系统疾病的嗜酸性粒细胞衍生神经毒素。
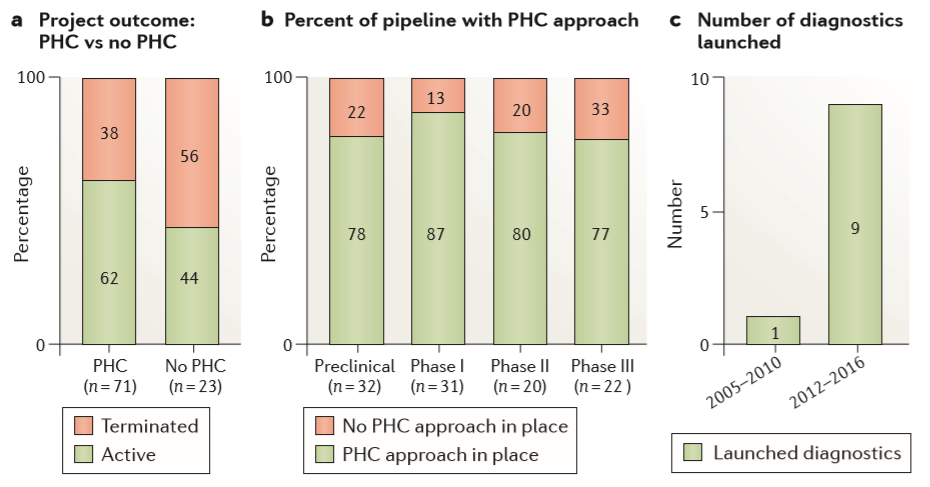
Figure 6. Impact of personalized health care on the portfolio
在整个阿斯利康管道中,2012年至2016年间的项目对选择前瞻性的患者有足够的理解,并且与进入下一个发展阶段的概率持续增加相关——有前瞻性选择的项目占比62%,没有患者选择策略的项目占44%(Figure 6a)。这创造了一个环境,在药物发现项目的早期,团队会考虑患者分层,从而能够及时开发患者选择性生物标志物,并在适当时开发辅助诊断。因此,从先导化合物优化开始,2012-2016年投资组合中约有80%项目具有患者选择性策略(Figure 6b),而2005-2010年的这一比例不到50%(REF. 7)。最后,关注定义“正确的患者”意味着我们在2012-2016年开展了九项辅助诊断测试,而2005-2010年仅开展了1项(Figure 6c)。
适配正确的商业潜力
根据我们的经验,在许多公司中,过分强调商业潜力可能会错误地推动项目。这在我们最初的2005-2010年评估中很明显,在该评估中,项目是基于商业信心而非科学信心而推进的,在中期和后期的开发中也毫无意外地失败了。
如今,在选择候选药物时,项目团队和管理委员花费相对较少的时间用于估算年销售或商业价值的峰值,而是专注于使用适当的临床比较工具生成有效性和安全性数据所需的内容,以增强人们对该项目能够提供差异化医疗服务的信心。在这一阶段,项目已经推出多年,因此商业估价最好是有根据的猜测,最坏是完全错误的猜测。因此,“正确的商业”模式在项目的临床生命早期并不支配管理或决策。然而,当一个项目达到III期投资决策时,团队需要有一个详细的商业评估,清楚地说明患者人数、未满足的医疗需求、与标准护所需的差异需求、全球偿付所需的付款人标准、竞争对手环境和可能的商业销售预测。还应注意的是,团队需要保持灵活性,并意识到不断变化的外部环境,因为决策标准可能需要根据该环境的变化进行变化。
创建正确的文化
把重点放在“求真”行为上,对项目提出“杀手”级问题,是我们文化变革的重要组成部分。我们通过加强定量科学和定量决策,继续推进“正确文化”主题。正如Donelan等人所指出的,投资决策通常是在竞争环境中存在高度科学不确定性的情况下做出的。此外,正如Owens和同事强调的那样,就什么将构成概念验证达成共识可能是一个挑战;科学家自然需要高水平的证明,对要测试的概念持有细微差异的观点,并且在进行做/还是不做(Go/No-Go)的III期投资之前,经常主张进行一个或多个确定的II期临床试验。
对处于早期开发阶段(从临床前开发到II期结束)中的每个分子应用先验定量决策标准是我们解决这些挑战的方法,并提高了在中期分析时作出决策的能力。虽然这些分析主要是徒劳的,但也可以使用适用于中期数据的决策标准来加速发展。我们采用的定量方法是从Lalonde等人首先描述的方法修改而来的,由Frewer等人改进,并且基于使用上、下置信区间来对观察结果的证据强度进行分类。该方法可根据研究应用于安全性和/或有效性的评估。例如,当测试患者的疗效时,将观察结果与较低的参考值和目标值进行比较。较低的参考值被定义为用于开发所研究的化合物的最小的临床有意义的治疗效果。典型地,它被设置成假设与护理标准一致的功效,但是还可以设置成假设在没有替代治疗的情况下或者在除了功效之外的差异可能足以用于进展的情况下没有差异。目标值通常设定在所需的效果水平,以便确认化合物可以作为新的治疗选择。这一决定框架也适用于计划的临时分析的结果。在这种情况下,可以预测在中间点做出”go”决策的概率,基于该预测做出无效或加速决策。例如,如果在研究结束时存在小于10%的”go”决定的预测概率,那么这可能是无效和早期”停止”的决策理由。如果在研究结束时有超过80%的”go”决定的预测概率,那么这可能是早期加速决策的理由。
在我们肿瘤投资组合中的一个应用实例是一项正在进行的单臂研究,该研究组合了AZD8186(PI3Kβ和PI3Kδ的抑制剂)和AZD2014(CREB调节转录共激活子1(TORC1)和TOTC2抑制剂)对三阴性乳腺癌患者的影响。目标应答率设为30%,最低应答率设为10%。当样本量设置为n=21时,观察到的应答率为24%或更高,这是一个明确的“继续”决定,14%或更低的响应率将导致一个明确的“停止”决定。14–24%范围内的响应率(琥珀色区域)将导致进一步检查附加的预定义终点,以做出决定(S3 Figure)。以这种方式常规地预先定义决策标准的好处是,需要足够的时间为目标和决策的最小值提供证据基础,并且决策以标准、透明的方式呈现。最后,当数据可用时,根据预先定义的决策标准快速做出决策,因此团队不会浪费时间对治理委员会进行二次猜测,也不会在期望的有效性信号搜索中挖掘数据。
关于生产率的总结
待续
Figure 7. Metrics for project costs, cycle times, publications and investor sentiment


