
摘要:本文探讨了主成分分析(PCA)在蛋白质分子动力学(MD)模拟和药物发现中的应用。MD技术在模拟复合物行为时非常有价值,但分析和呈现结果非常具有挑战性。PCA作为一种降维技术,能够帮助从MD数据中提取有用信息,并评估蛋白质的构象空间。本文通过一个具体的例子说明,使用PCA可以将MD模拟中的3D坐标转换为线性正交向量,即主成分(PC),从而在2D图中对结果进行视觉分析。本文还讨论了如何通过PCA揭示蛋白质动力学模拟中隐藏的模式,并展示了如何利用PCA分析不同条件下蛋白质的构象采样。此外,还演示了如何在FEP实验中用PCA识别离群值。在即将发布的Flare V9中包含分子动力学轨迹的PCA分析功能。
作者:Federico Issoglio/2024-07-18
编译:肖高铿
概述
在药物发现中,蛋白-配体相互作用的详细知识是理解两个分子之间识别过程的关键,而分子动力学(MD)等技术在使用原子细节模拟复合物的行为时很有价值。几个方面可以影响蛋白的动力学:突变,不同配体的结合,质子化状态(可滴定的残基或配体),脂质双层的组成(在膜蛋白的情况下)和温度,这里仅举几例。这些也会同样会影响复合物的稳定性。
通过MD结果的分析和呈现以帮助指导项目具有挑战性。当需要比较几种MD时,尤其是在大型的自由能微扰(Free Energy Perturbations,FEP)实验中,尤其如此。一种常见的做法是使用主成分分析(Principal Component Analysis,PCA)来降低数据的维度,同时保留最重要的信息。这种分析经常用于评估蛋白质可以进入的构象空间,与其他常用的分析工具(如RMSD,RMSF和回转半径等等)相比,它可提供更多的细节。正如下面所举的例子,从PCA数据中提取有用信息在很大程度上依赖于适当的可视化工具使用。
用PCA图对结果进行视觉分析
我们可以使用PCA的视觉分析来更好地了解MD模拟所访问的构象。使用PCA,我们可以将轨迹文件中所有帧的3D坐标转换为一组线性正交向量,称为主成分(Principal Components,PC)。为了在一个简单的体系中描述这一点,在图1中,每个蓝点描述了轨迹文件中每一步一个原子的x和y坐标,红色箭头代表该体系的前两个PC。
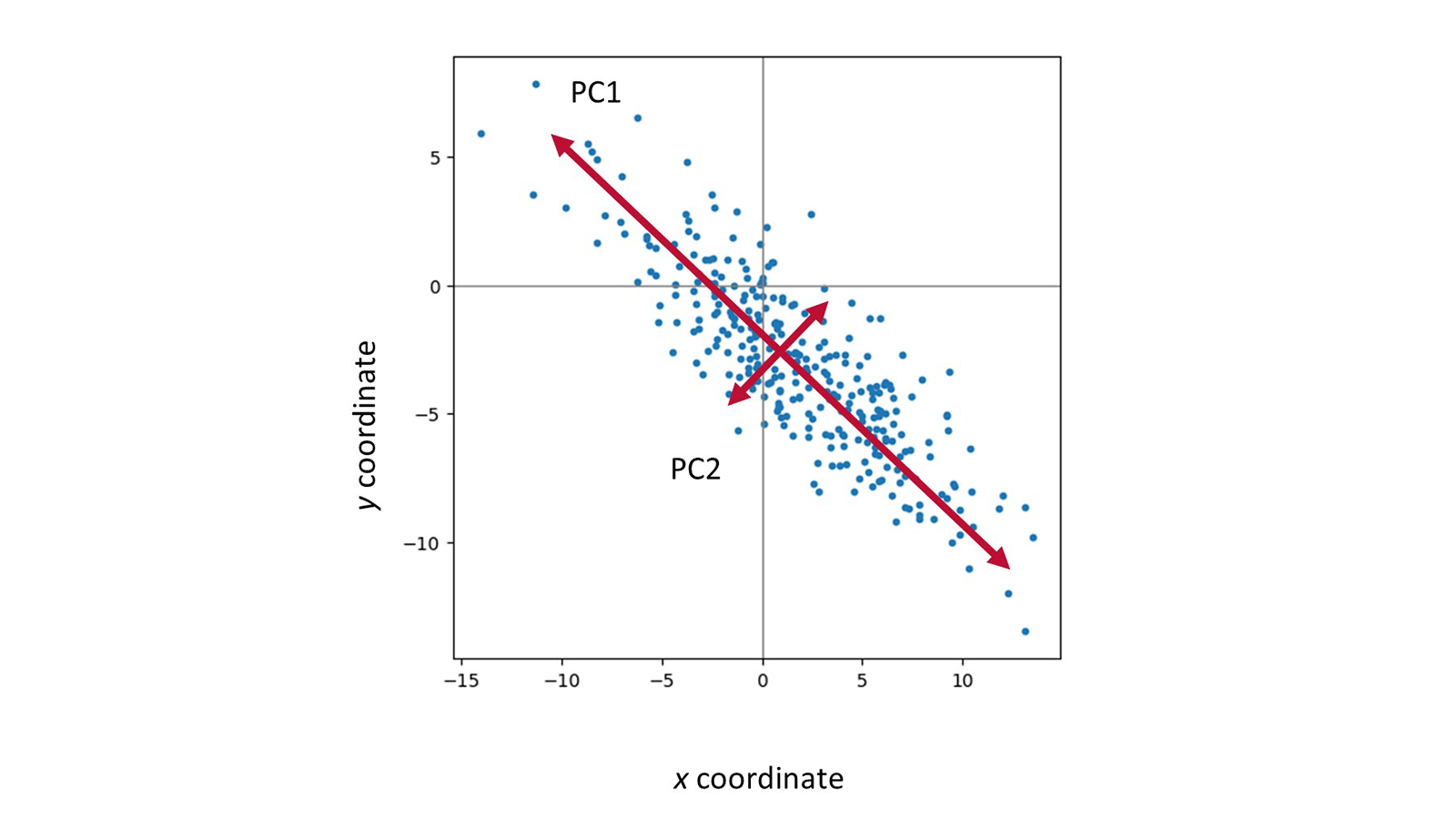
图1. 单原子体系(蓝点)x和y坐标的表示,以及从PCA获得的相应前2个特征向量的定性表示。
现在,让我们想象一个三维体系(例如蛋白质分子动力学模拟),在分析中我们假设得到了一个结果,其中我们仅用1个PC即可用来解释100%的变量:这意味着所有原子的运动集合可以表示为一个主成分(PC)的线性变换(即刚性蛋白质的平移)。在一个更真实的例子中,如果前两个PC所可解释的变量信息分别为52%和29%,那么可以在2D图中跟踪这两个PC的演变来解释81%的蛋白质动力学(图2B)。
严格地说,为了完成这种分析,使用3D坐标生成3N x 3N的协方差矩阵(其中N是原子数),并且从该矩阵的对角化我们获得特征值(每个原子的每个坐标的方差),PC是相应的特征向量(按方差的降序排列)。通过将原始3D坐标投影在前两个PC上,我们生成了一个代表蛋白质动力学的2D图,正如在采样到的构象上发现的两个最大的不相关运动所描述的。这意味着,例如,我们可以确定蛋白质是否在模拟时间内实现了构象收敛,比较不同的条件,或者确认平衡阶段是否正确设置。
用PCA图揭示在蛋白动力学模拟中隐藏的模式
为了在实际示例中说明此分析,使用RMSD和PCA分析了50ns的MD模拟(图2)。根据RMSD分析(图2-A),在模拟时间10、30和45ns处的构象将是等效的(橙色星型)。在PCA上,给出了前两个PC的2D图,我们可以使用色条跟踪模拟时间(图2-B)。类似地,在模拟时间10、30和45ns采样的构象用白色星型标记。我们可以看到,PCA表明这些构象并不等同,从40到50ns范围内采集的构象空间取得结构上的收敛。换句话说,在这个例子中,从PCA图获得的证据表明蛋白质探索到了3个宏观状态,而RMSD没有捕获到这一点。
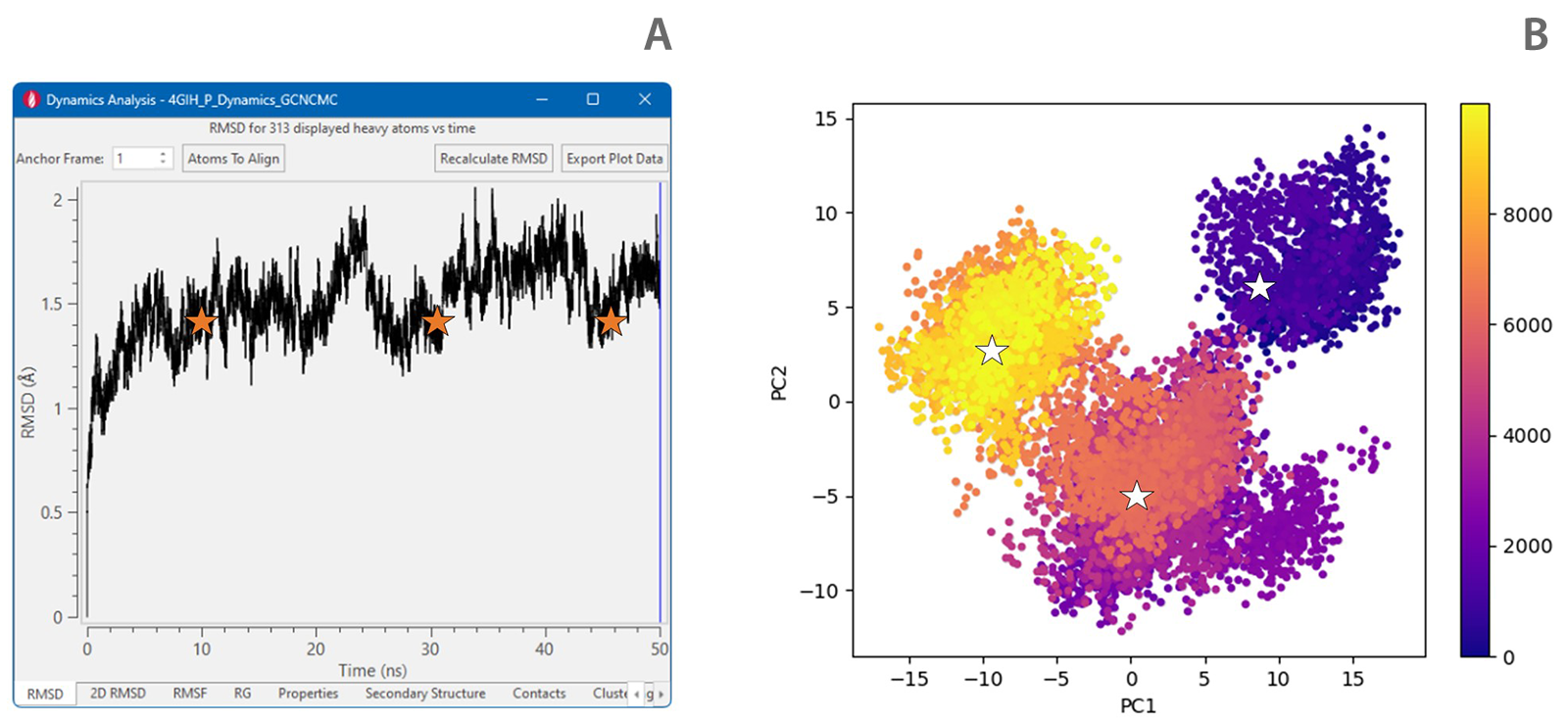
图2. A)RMSD分析:从这个图中,我们没有强有力的证据表明该蛋白质在10到50ns的模拟时间内呈现出不同的构象。B)通过将蛋白质轨迹的坐标投影到第一和第二特征向量(PC1和PC2)上,可以获得相同体系的PCA结果。数据点根据模拟时间(以帧为单位)进行着色,从而可以跟踪模拟过程中采样构象的演变。在这个图中,我们获得了关于蛋白质采样的构象空间的更全面的信息(即,在这个例子中,证据表明蛋白质探索到了3个宏观状态)。白色星型表示与A图中橙色星型相同的模拟时间。
从认识我们的体系到选择新的候选化合物
随着计算能力的提高,使用MD评估结合的候选药物分子的稳定性已成为常见做法,在这个场景里,我们将用PCA显示不同情况如何对采样的蛋白质构象产生重大影响。在图3中,在不同条件下评估相同蛋白质的完整PCA分析中使用的八个轨迹的投影被描绘为灰点,而其中两个轨迹的单个结果在模拟时间后被着色(与图2-B相同)。在这个例子中,我们分析了的相同二聚体蛋白在两个活性位点都被占据的共晶复合物(图3-A)或第二个结合位点未被占据的共结晶复合物(图3-B)获得的结果之间的差异。在两种情况下对占据的活性位点的蛋白质残基位置都进行PCA。PCA图表明,在图3-A中探索到的构象空间更窄、更接近初始构象,而在图3-B中显示的情况下,蛋白质正在经历值得注意的重组。因此,我们可以得出结论,变构效应连接了来自两个亚基的活性位点,PC1和PC2获得的显著不同一组数值证明了这一点。
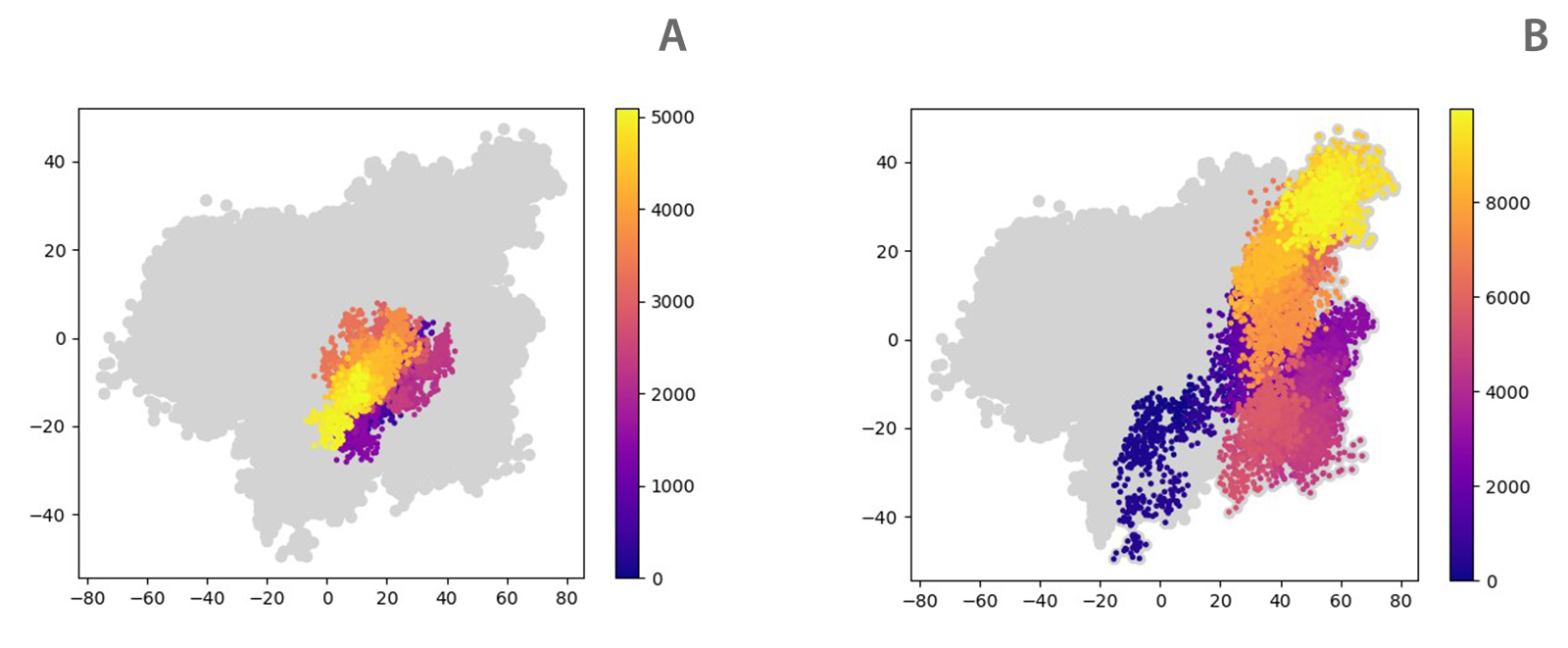
图3. 在两个不同的200ns MD中跟踪二聚体蛋白的构象采样,使用一个结合位点的所有重原子3D坐标进行PCA。A)轨迹包括两个启动子活性位点中的共结晶化合物。B)从MD模拟获得的轨迹,其中只有一个结合位点被占据。在两幅图像中,每种情况的数据点都根据模拟时间(以帧为单位)进行着色,并且在PCA计算中使用的所有八个轨迹(包括二聚体的不同条件)探索到的空间用灰点描绘。
PCA可以用来支持FEP实验以识别同系物中的离群值。如图4所示,我们分析了包含28个配体结构的FEP研究数据。对于每个复合物,将每个轨迹的最后一帧投影到PC图(洋红色圆圈,图4-B)上,该PC图由靶标和参比先导化合物复合物的初始200ns MD模拟定义(图4-A)。从事这项工作的科学家可以使用此分析来识别PC1和PC2描述的离群结构,并评估观察到的差异的来源(例如,诱导拟合效应,非收敛结果,由于初始叠合不良引起的构象噪声等)。在这个例子中,四个配体显示较大偏差(图4-B,绿色椭圆形),它们要么具有不止一个取代位点,要么含有空间体积大的R-基团取代基。
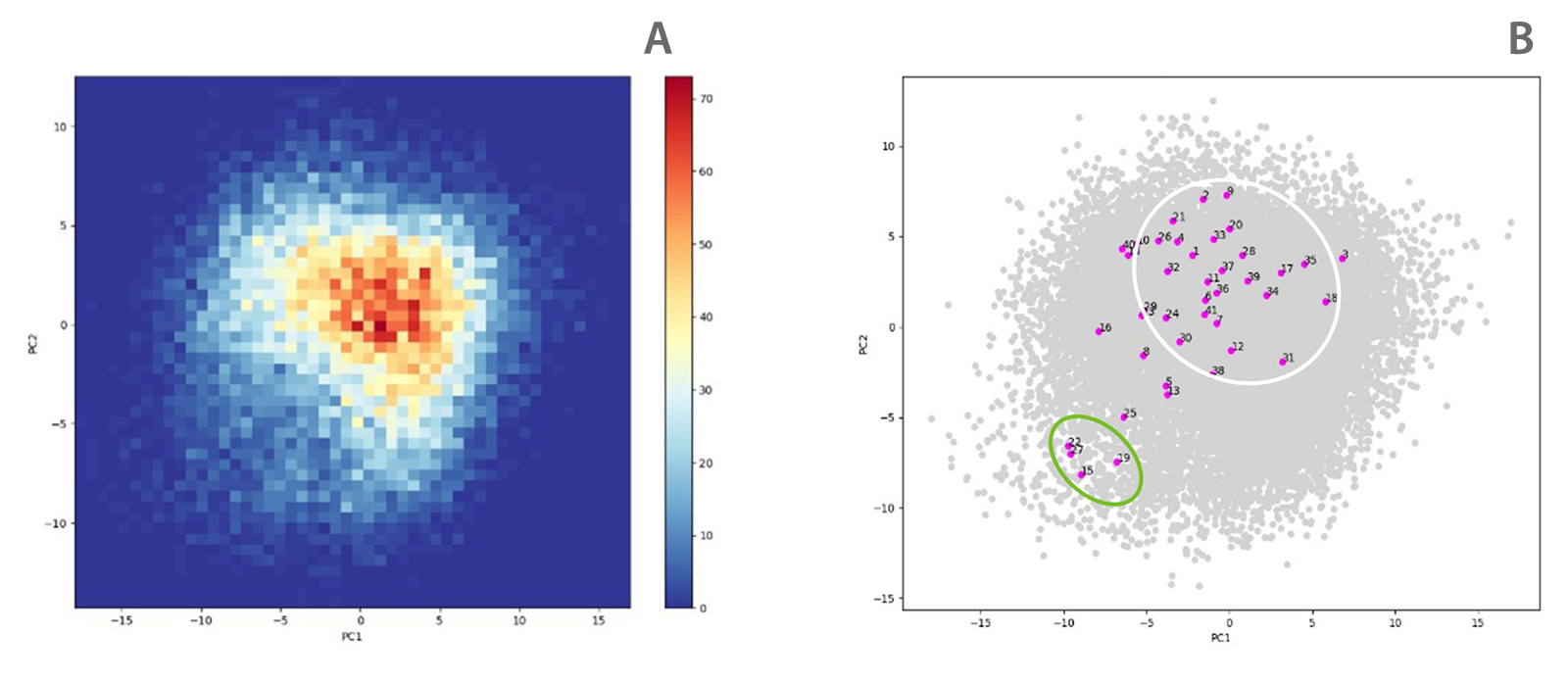
图4. A)使用MD获得前两个PC生成的热图作为参比(FEP计算前一步),以及从每个变换最后一帧获得的41个FEP轨迹结构。B) 覆盖的构象空间都用灰色表示,从FEP获得的结构投影用洋红色表示。对应于热图(A)中丰度最高的区域用白色椭圆形(B)分隔。
在Flare™内可视化PCA分析
在Flare中,我们提供了各种各样的工具来分析MD轨迹结果,利用pyflare可以显著地扩展其分析能力。Pyflare支持在Flare GUI中编辑、运行python脚本。对于这里介绍的分析,我们将面向对象的python工具包MDAnalysis1-2与Cresset编写的pyflare脚本相结合。如果您对使用这些脚本进行研究感兴趣,请与我们联系,我们的支持团队将很乐意提供这些脚本。
分子动力学轨迹的PCA分析即将在Flare V9中与大家见面,请继续关注我们的发布公告!
文献
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf, and O. Beckstein. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32 (2011), 2319–2327. doi:10.1002/jcc.21787
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, D. L. Dotson, J. Domanski, S. Buchoux, I. M. Kenney, and O. Beckstein. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In S. Benthall and S. Rostrup, editors, Proceedings of the 15th Python in Science Conference, pages 98-105, Austin, TX, 2016. SciPy. doi:10.25080/Majora-629e541a-00e


