
摘要:Ligandscout是非常专业、易用的药物设计平台, 除了药效团建模与虚拟筛选外,还整合了分子对接软件AutoDock与AutoDock Vina。本教程演示了四种对接方式:(1)从配体-受体复合物结构出发进行分子对接;(2)blind docking:仅有受体结构、结合位点未知条件下盲法对接; (3)自定义结合位点进行对接计算;(4) Ligandscout预测结合位点进行对接计算。
作者:肖高铿
联系:gkxiao@molcalx.com
一. Ligandscout与分子对接软件VINA
LigandScout是新兴的、非常专业、易用的药物设计平台,每年使用Ligandscout进行药物研究的文章也越来越多。AutoDock V4.2与AutoDock Vina是两个非常著名的分子对接软件。
LigandScout开发团队的目标之一是提供最简单、易用以及可靠的药物设计平台,在最新的LigandScout 4里整合了AutoDock4.2与AutoDock Vina这两个分子对接软件。本视频教程的目的是为大家演示如何用LigandScout进行分子对接计算。
关于AutoDock与AutoDock Vina
| 分子对接软件 | 版本 | 授权方式 | 官网 |
| AutoDock | 4.2.6 | GNU license | http://autodock.scripps.edu |
| AutoDock Vina | 1.1.2 | Apache license | http://vina.scripps.edu |
无论是AutoDock还是AutoDock Vina,在Ligandscout平台上都支持下面四种对接方式:
- 从一个配体-受体复合物晶体结构出发:共晶配体所在的区域为结合位点进行对接计算,预测结合模式
- Blind Docking:仅有蛋白晶体结构而没有共晶配体,不知道结合位点在哪里,让化合物在整个蛋白区域漫游对接,预测可能的结合模式;
- 通过氨基酸残基来定义结合位点进行对接计算,预测可能的结合模式;
- 还可以通过Ligandscout预测结合位点进行分子对接计算,预测可能的结合模式;
二. Ligandscout VINA分子对接计算操作步骤
2.1 从一个配体-受体复合物晶体结构出发, 以配体所在的结合位点为口袋进行对接计算
以CDK2激酶及其抑制剂的复合物晶体结构为例(PDB code:1KE6),包含了下面的操作过程:
- 在PDB 4-字母框里键入蛋白质复合物结构的PDB代码:1KE6
- 选择对接计算的结合位点:点击黄色盒子即可;
- 导入待计算的化合物结构,支持多种文件格式,包括常见的SMILES、SDF与mol2格式等: File >Insert>选择要计算的化合物
- 对接计算:Molecule>Dock Ligand> AutoDock Vina
导入后,在左上角你会发现有Core Molecule, Injected Library Molecules与MacroMolecule,点击Core Molecule旁边的眼睛,让它变灰色,以便在docking的时候只有Injected Library Molecules被docking。
如果你导入了多个分子,可以用鼠标在library的分子表单在Name列点击对应化合物的就可以选择要对接的化合物。
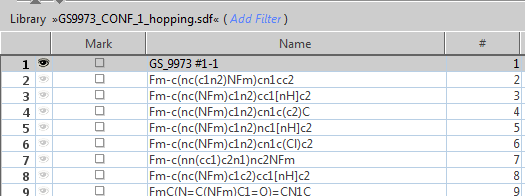
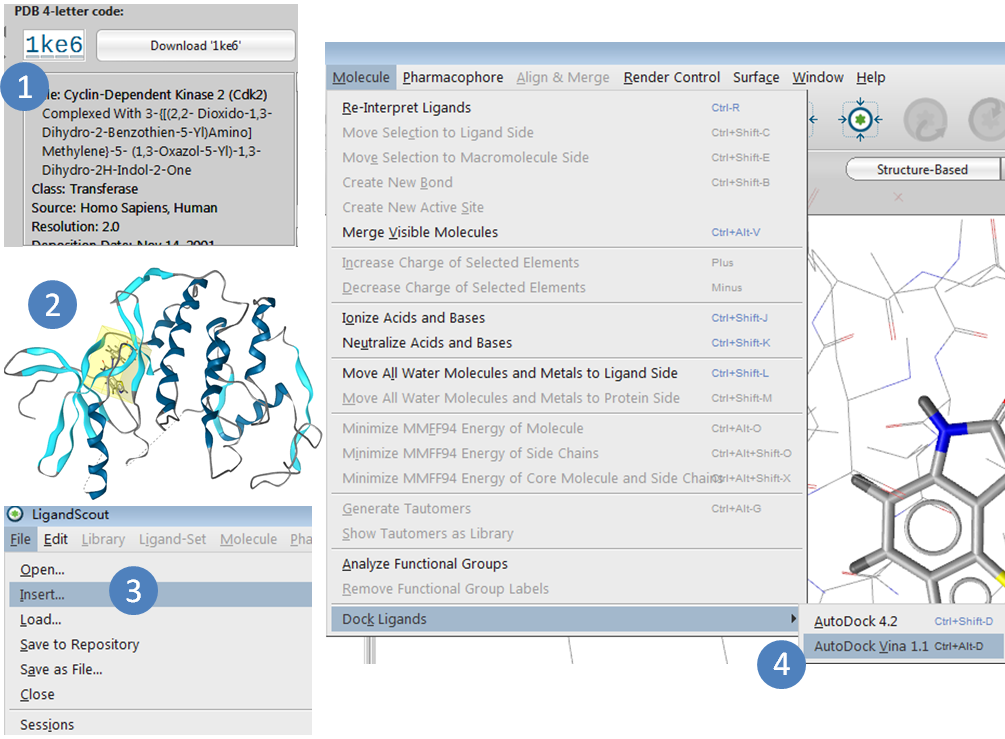
图1,Ligandscout教程–Vina分子对接操作步骤
2.2 Blind Docking: 结合位点未知让配体在整个蛋白漫游定位
以ABL激酶及其抑制剂STI(Gleevec, 格列卫)的复合物晶体结构为例(PDB code:1IEP),删去配体,并让对接计算的区域包含了整个蛋白结构,包含了下面的操作过程:
- 在PDB 4-字母框里键入蛋白质复合物结构的PDB代码1IEP,点击Download开始下载
- 删除B链,全部的水分子以及全部HETATM原子(包含的共晶化合物),仅留下ABL激酶的A链
- 设定整个蛋白为对接的区域,以便进行blind docking
- 导入待计算的化合物结构
- 对接计算
如果之前已经下载过1IEP,现在会提示“renew 1iep”,点击“Renew 1IEP”按钮。
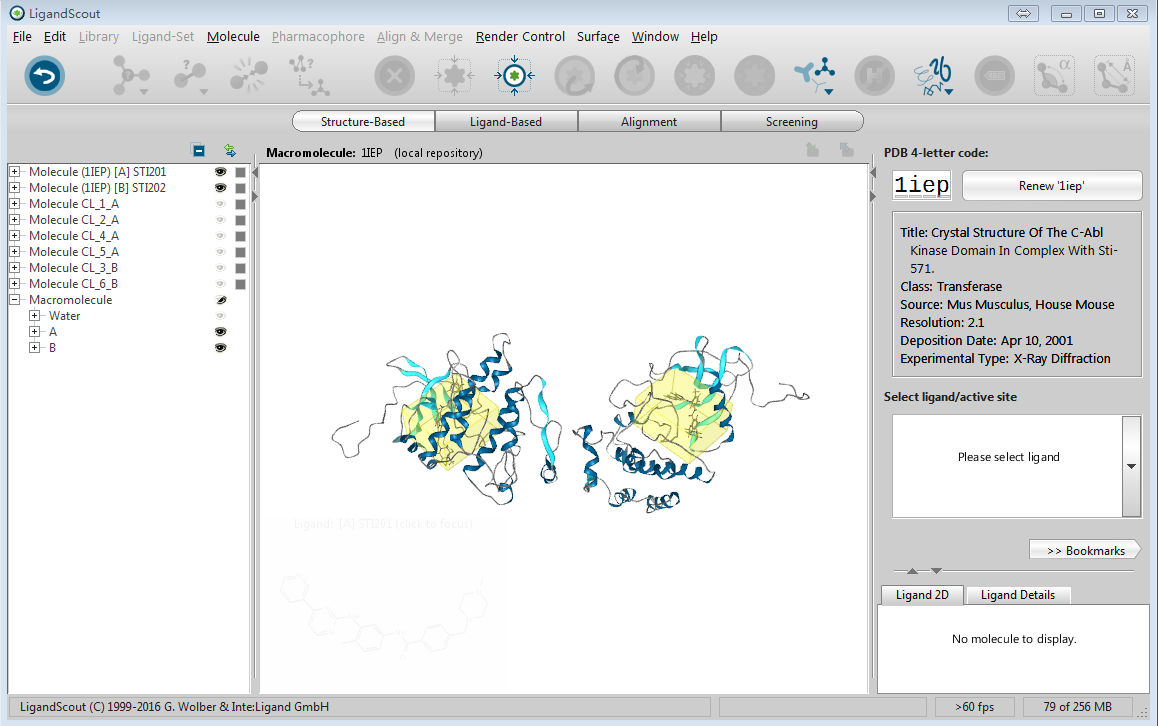
图2. 下载ABL激酶与抑制剂STI571复合物结构(PDB Code: 1IEP)
用安装Ctrl键,用鼠标单击需要删除的成分STI、Cl原子、水与B链,仅留下Macromolecule栏下的A未被选中;再按Delte按钮,删除被选中的元素,或者使用菜单命令:Edit>Delete Selected Element
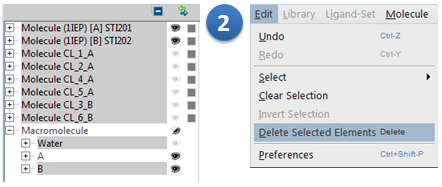
此时左侧的分子导航栏其它元素都不见了,只剩下Macromolecule。
注意:删除Chain B是个学习“删除”操作的练习,不是必须的步骤。因为在Ligandscout里,你可以直接选择你感兴趣的区域,再Create new active site就可以了。
(1)选中整个大分子:
用鼠标单击分子导航栏的Macromolecule。
(2)设定结合位点覆盖整个蛋白:
Ligandscout菜单栏:Molecule>Create new active site;
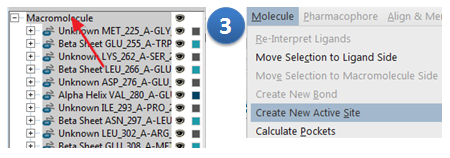
活性位点创建完毕,会发现一个黄色的盒子覆盖住整个ABL结构,如图3所示。
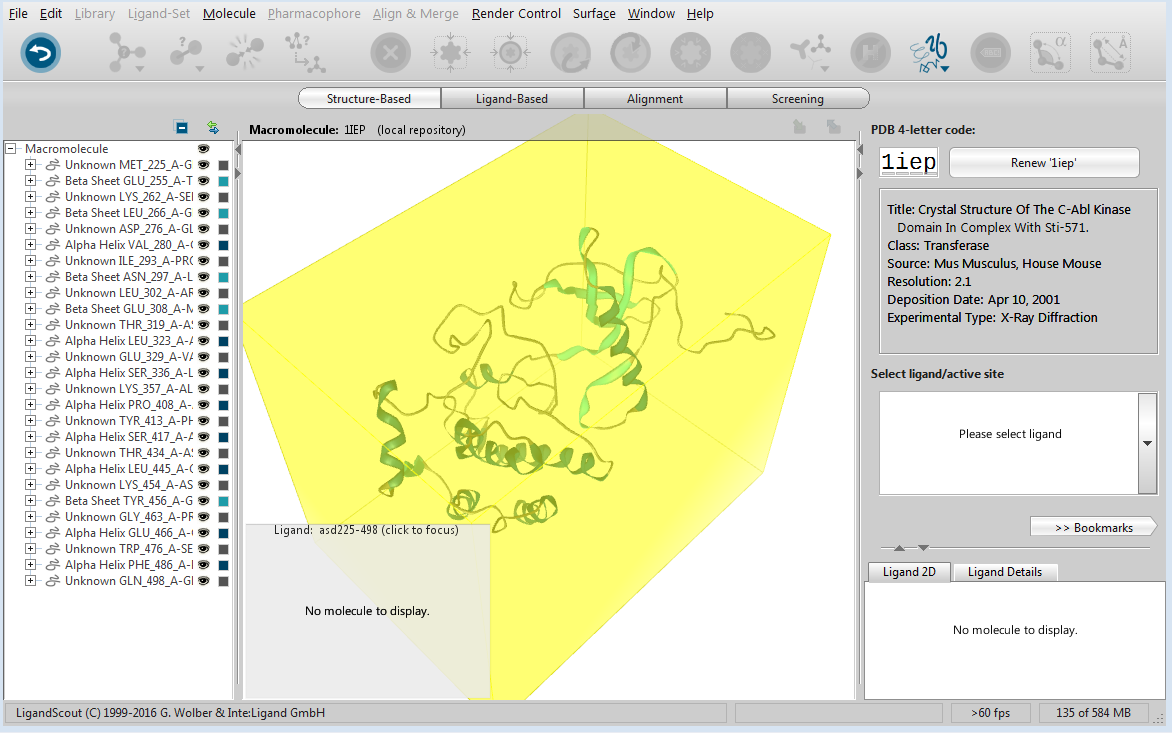
图3, 创建一个新的结合位点覆盖整个ABL结构进行Blind Docking
(3)单击黄色口袋,确认结合位点
单击黄色口袋,确认结合为点,这时已经准备好分子对接的所需的受体结构准备工作。
Ligandscout菜单栏: File >Insert>选择要计算的化合物
Ligandscout支持多种文件格式,包括常见的SMILES、SDF与mol2格式等。以STI结构为例:
1 | CC1=C(C=C(C=C1)NC(=O)C2=CC=C(C=C2)CN3CCN(CC3)C)NC4=NC=CC(=N4)C5=CN=CC=C5 gleevec |
将上面行保存为gleevec.smi,再导入到Ligandscout。Ligandscout自带的iCon会为化合物生成优质的3D结构用于分子对接,结果见图4,现在用于对接的配体结构也准备好了。
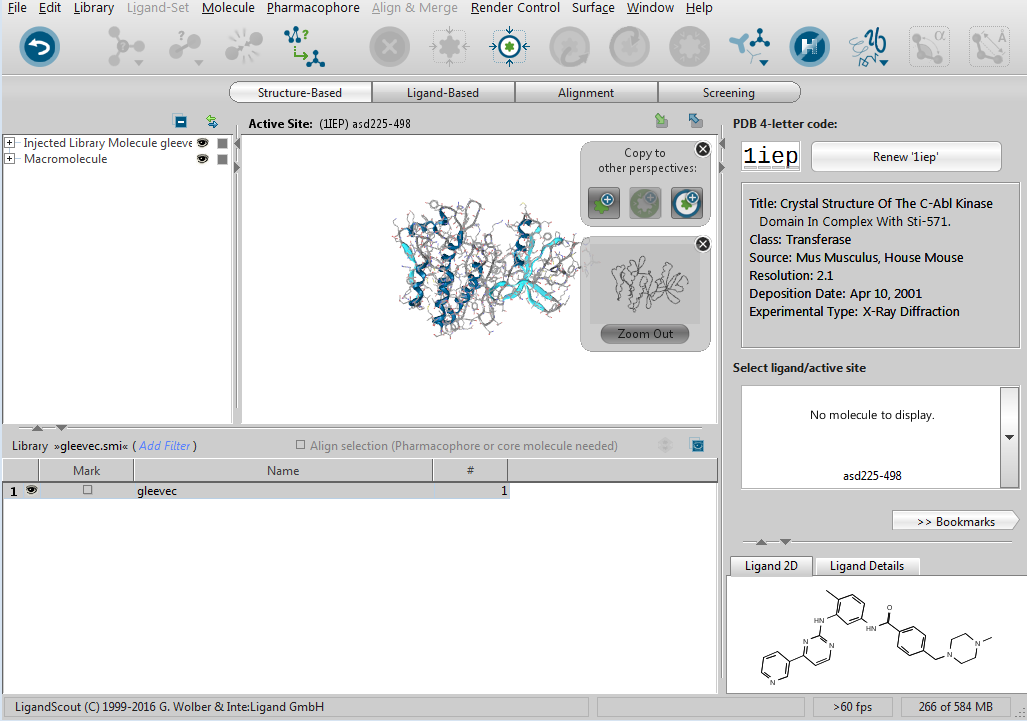
图4, 导入Gleevec准备对接计算
按组合键”Ctrl + Alt D”开始设置AutoDock Vina参数进行对接;按组合键”Ctrl+Shift D”开始设置AutoDock参数进行对接计算。或者通过菜单 Molecule>Dock Ligand> AutoDock Vina开始对接计算
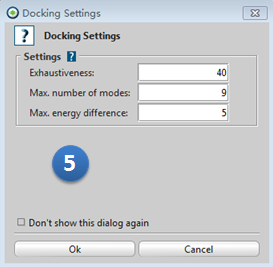
图5, 对接参数设定
Exhaustiveness: 全局搜索的程度,越大计算时间越长,对于大的柔性分子有必要加大该值提高对接精度
Max. number of mode: 输出结合模式的最大数量
Max. Energy difference: 输出pose的能量与最低的那个比较差值的最大值
点击OK开始计算
2.3 自定义结合位点对接计算: 通过氨基酸残基来定义结合位点进行对接计算
操作步骤与2.2的blind docking相同,不同的之处在于第3步的设定结合位点:不是单击Macromolecule,而是展开Macromolecule,单击构成活性位点的氨基酸残基(按住Ctrl键,单击多个残基)。
2.4 预测结合位点进行对接计算
操作步骤与2.2的blind docking相同,不同的之处在于第3步的设定结合位点。
- 开始预测结合位点
- 建立结合口袋
- 点击黄色盒子,确认对接的结合位点
ligandscout菜单:Molecule>Calculate Pockets
Ligandscout的grid会预测可能的结合位点,结果如图6所示:
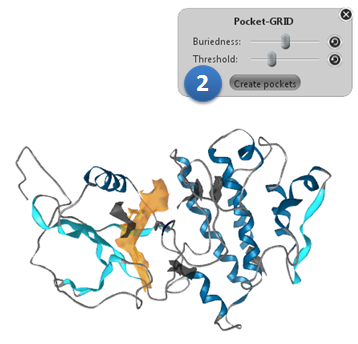
图6, Ligandscout的结合位点预测:通过调整阈值,可以调节口袋的大小
点击Create Pocket生成结合位点,见图7,此时会出现一个盒子包围着一个结合空腔。
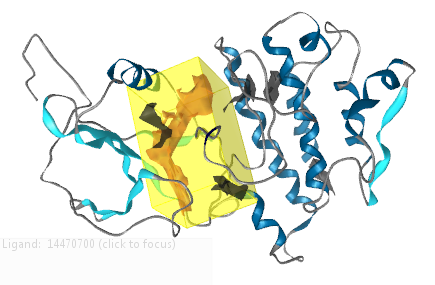
图7, 结合口袋的创建
点击黄色盒子,确认对接的结合位点,准备好对接计算。
三. 分子对接结果分析
1. pose浏览
对接计算完毕后,在分子表单中列出对接的pose,每个pose一行,在name列点击任意一行,则3D视窗区与2D视窗去则分别以3D与2D的方式展示配体与受体的相互作用模式,见图8。
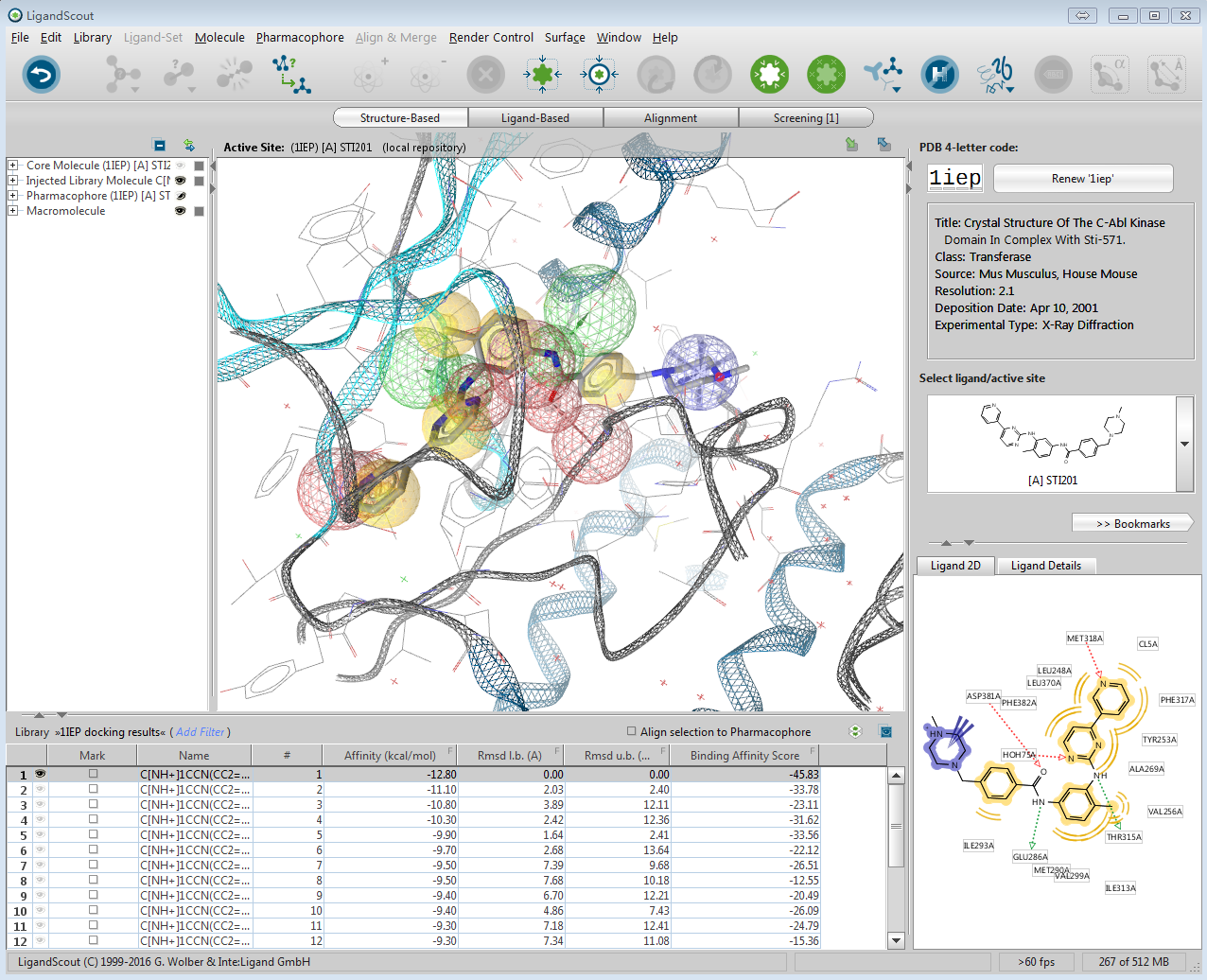
图8, 结果浏览
选择一个pose,点击“create Pharmacophore”按钮,则生成该pose与蛋白之间的药效团。
2. 3D或2D相互作用模式图片的保存
File> Save as Files出现Save File对话框:将输出设置为图片格式,比如PNG格式;选择要保存的内容,2D或3D,设置图片分辨率(支持任意大的分辨率),点击保存,见图9。
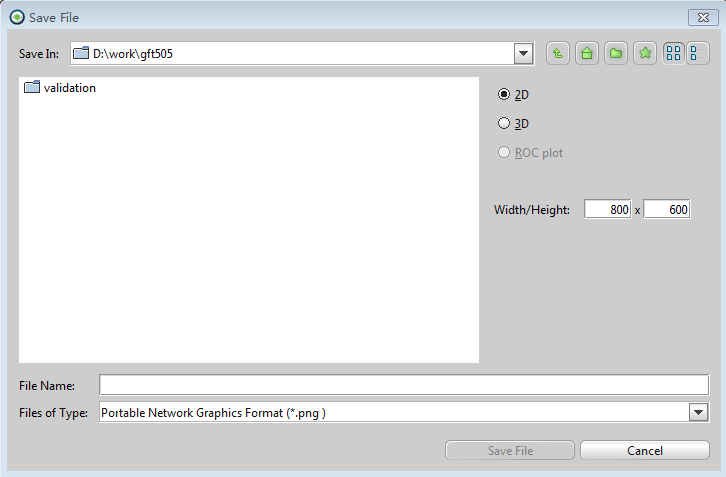
图9, 2D或3D相互作用保存为图片
四. Ligandscout分子对接演示
五. 从这里开始还可以做什么
1.用分子形状给对接的pose打分;
2.给对接的pose用结合亲合力打分函数进行打分,分值越低,表明该pose的结合亲合力越大;
3.用药效团对pose打分;
4.对pose生成药效团模型,观察结合模式:支持2D、3D展示;
5.基于药效团的虚拟筛选;
6.用FreeForm测试结合构象稳定性,排除假阳性。
六,联系我们,获取一个月的免费试用
电邮:info@molcalx.com
电话:020-38261356
单位:广州市墨灵格信息科技有限公司
网站:http://www.molcalx.com.cn/ligandscout


