
摘要:本文演示了如何用Pymol与Openbabel从蛋白复合物结构出发准备AutoDock Vina计算所需的蛋白结构文件、配体文件、对接参数文件,并进行VINA分子对接计算。本文获得的这些文件,可进一步用于Cloudam VirtualFlow的高通量虚拟使用。
肖高铿/2020/11/05
特别说明(2021-12-04)
鉴于用Openbabel 3.1以及更早版本转化为PDBQT格式时存在柔性键错误定义的情况,在这个问题得到修复之前,我不推荐用基于OpenBabel进行格式转化的Vina对接计算。
前言
抗白血病药物格列卫是ABL激酶抑制剂,1IEP晶体结构为格列卫(STI, GLEEVEC)与ABL激酶的复合物结构。在本练习中,演示了用Pymol从1IEP复合物结构出发准备起始的受体结构文件,并用在云E算力平台上预装的OpenBabel与分子对接软件AutoDock VINA完成:1)蛋白结构准备;2)配体结构准备;3)分子对接参数设置;4)分子对接;5)结果文件的格式转化;6)可视化分析。本文准备的文件可进一步用于Virtualflow进行高通量虚拟筛选。
本教程的目的
本教程的主要目的是初步了解:
- 如何用PYMOL准备蛋白结构
- 如何用OpenBabel转化PDB格式为PDBQT
- 如何准备Vina的对接参数文件
- 如何提交一个简答的VINA对接计算
用到的软件
- PYMOL
- OpenBabel
- Vina家族软件
用于蛋白的准备,需要预先安装于本地机器上。
Pymol的获取:https://github.com/schrodinger/pymol-open-source
用于格式转化,源代码与使用手册:http://openbabel.org。
OpenBabel已经预装于Cloudam云E算力平台,用module命令加载:
1 2 3 | module add Anaconda3/2020.02 source activate conda activate rdkit |
分子对接计算引擎VINA已经预装于Cloudam云E算力平台的virtualflow,可以复制到本地使用:
1 2 | export VFVS_ROOT=/public/software/.local/easybuild/software/virtualflow cp -r $VFVS_ROOT/vfvs/tools . |
更简单的办法是通过PATH直接使用:
1 2 | export VFVS_ROOT=/public/software/.local/easybuild/software/virtualflow export PATH=$VFVS_ROOT/vfvs/tools/bin:$PATH |
Vina家族对接软件可执行文件都在VFTools/bin目录里。更多的AutoDock软件包介绍见:https://ccsb.scripps.edu/autodock以及Stefano等人[1]的使用教程。
本教程需要了解的预备技能
- 对Linux的shell命令有初步了解
- 对分子可视化软件Pymol有初步了解
操作步骤
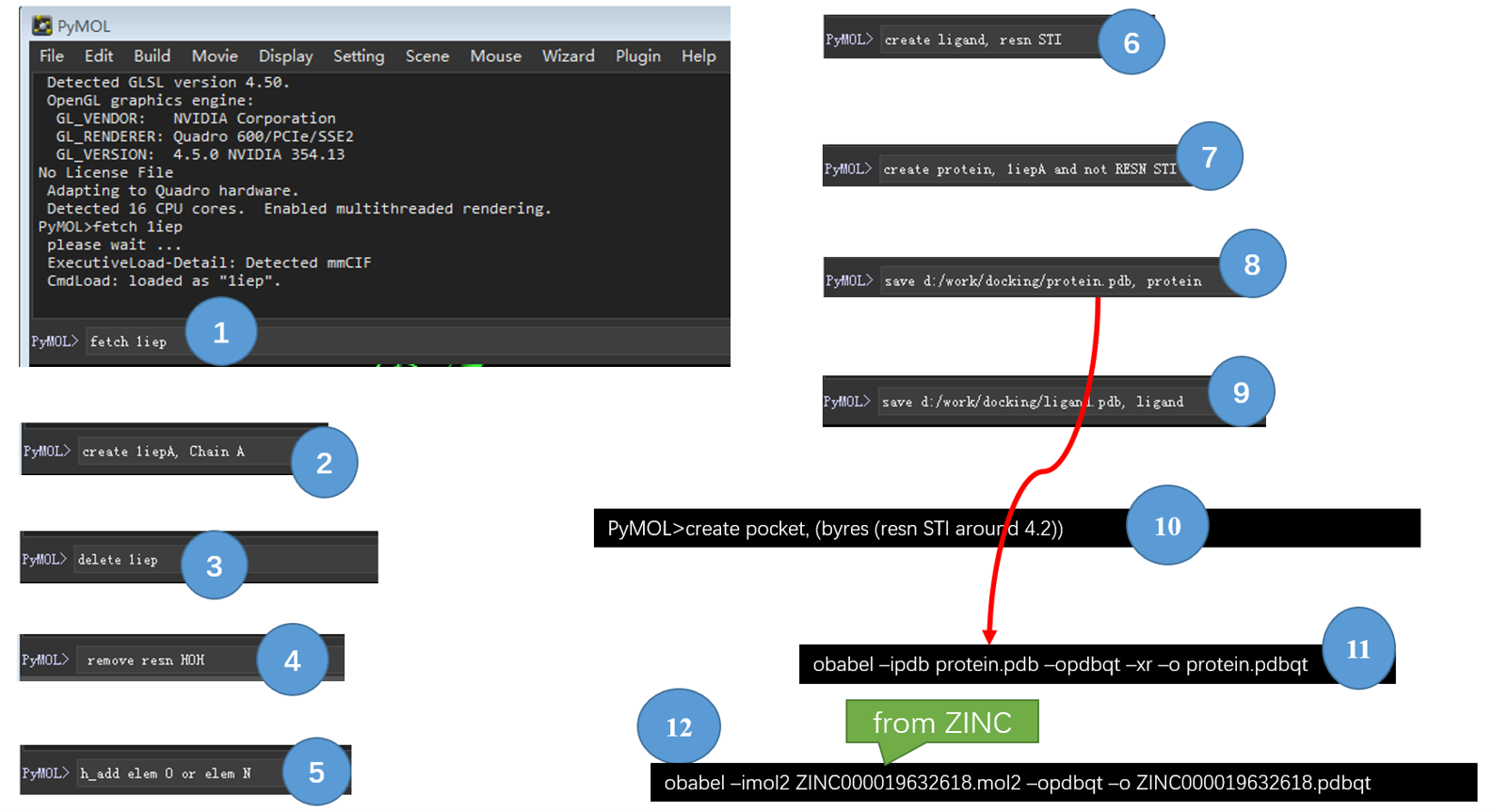
Figure 1. 用Pymol与OpenBabel进行结构准备的操作流程
1. 下载蛋白
在Pymol命令行键入:
1 | fetch 1iep |
2. 将蛋白A链单独保存
由于1IEP有两个链,我们仅需要其中一个(Chain A),在Pymol命令行键入:
1 | create 1iepA, Chain A |
3. 删除一个对象
为了操作简单,删除掉一个对象:
1 | delete 1iep |
4. 删除水分子
1 | remove resn HOH |
5. 加极性氢
1 | h_add elem O or elem N |
6. 将复合物的配体复制为一份单独的对象
1 | create ligand,RESN STI |
7. 将复合物的蛋白复制为一份单独的对象
1 | create protein, 1iepA and not RESN STI |
8. 保存蛋白为本地PDB文件
1 | save d:/work/docking/protein.pdb protein |
9. 保存配体为本地PDB文件
1 | save d:/work/docking/ligand.pdb ligand |
10.将配体(STI)周围4.2A残基定义为结合位点并创建对象
1 | create pocket, (byres (RESN STI round 4.2)) |
该操作的目的是可以对结合位点进行更好的渲染,不是必须的。
11. 将受体蛋白PDB格式转为PDBQT格式
本步骤将将第8步保存的受体文件protein.pdb转为PDBQT文件,用于后续分子对接计算用。
1 | obabel -ipdb protein.pdb -opdbqt -xr -O protein.pdbqt |
12. 将配体分子格式转化为PDBQT格式
以ZINC下载的mol2文件为例(在实际研究中,可能是其它软件比如chem3d保存的一个sdf或mol2文件),用openbabel转为配体用的PDBQT文件,用于后续分子对接计算用。
1 | obabel -imol2 ZINC000019632618.mol2 –opdbqt -O ZINC000019632618.pdbqt |
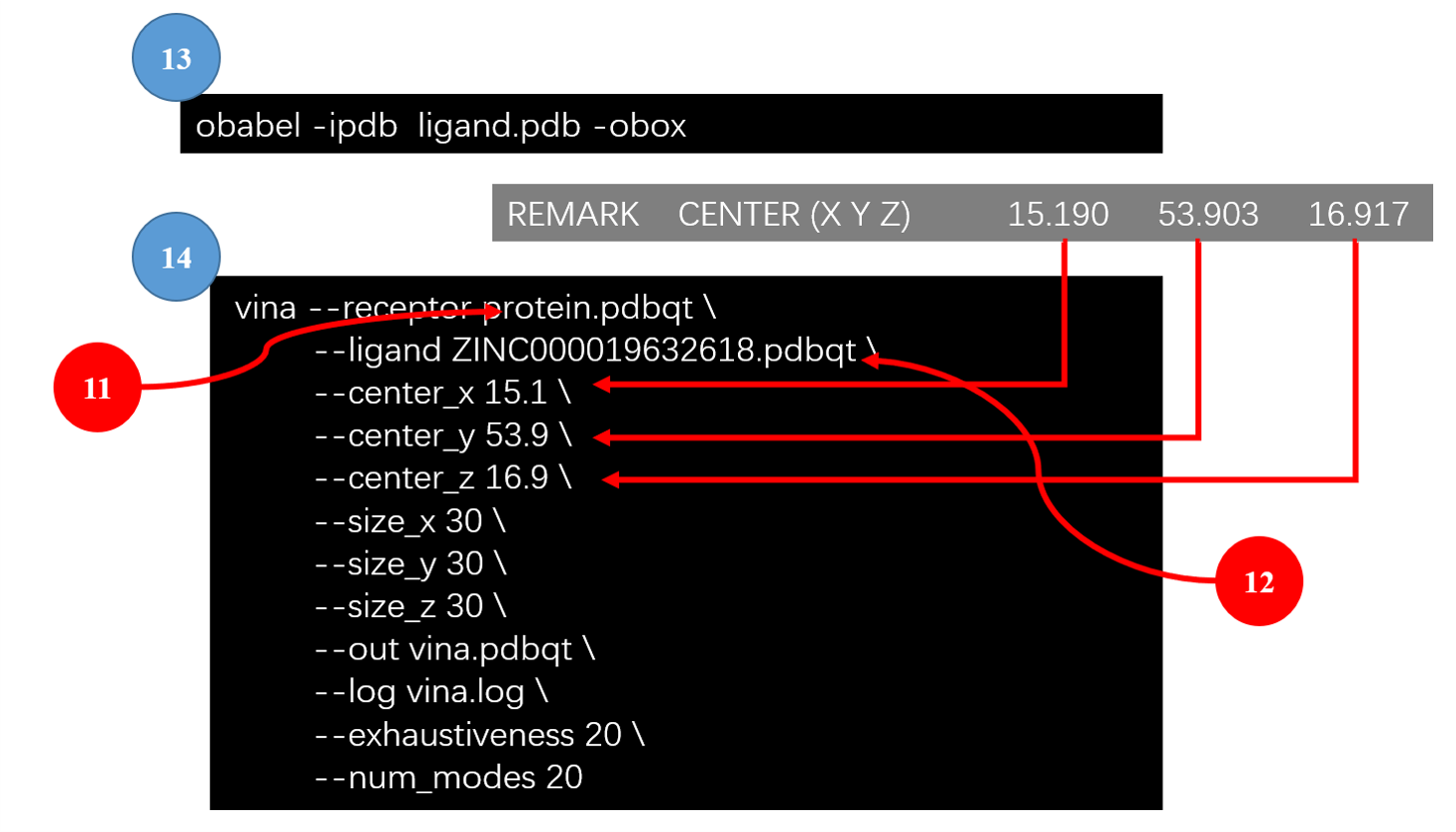
Figure 2. 对接参数文件的准备
注意OpenBabel对PDBQT的分支或可旋转键的定义可能会发生错误,以式1化合物为例来说明。
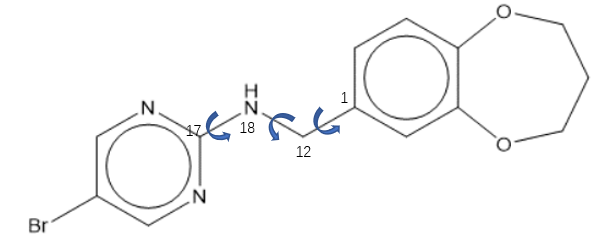
式1化合物,具有三个柔性键
lig_90.sdf是它的一个初始3D结构,用OpenBabel 3进行格式转化:
1 | obabel -isdf lig_90.sdf –opdbqt -O lig_90_ob.pdbqt |
会发现,Openbabel生成的pdbqt文件(lig_90_ob.pdbqt)仅包含两个可旋转键:C1_C12与C12_N18,具体见PDBQT文件的第4行与第5行。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 | REMARK Name = lig_90 REMARK 2 active torsions: REMARK status: ('A' for Active; 'I' for Inactive) REMARK 1 A between atoms: C_1 and C_12 REMARK 2 A between atoms: C_12 and N_18 REMARK x y z vdW Elec q Type REMARK _______ _______ _______ _____ _____ ______ ____ ROOT ATOM 1 C UNL 1 -6.646 -0.766 0.406 0.00 0.00 -0.027 A ATOM 2 C UNL 1 -7.226 0.465 0.208 0.00 0.00 +0.009 A ATOM 3 C UNL 1 -8.581 0.631 0.388 0.00 0.00 +0.050 A ATOM 4 C UNL 1 -9.389 -0.424 0.758 0.00 0.00 +0.162 A ATOM 5 O UNL 1 -10.699 -0.156 0.932 0.00 0.00 -0.488 OA ATOM 6 C UNL 1 -11.436 -0.874 1.946 0.00 0.00 +0.232 C ATOM 7 C UNL 1 -11.914 -2.258 1.457 0.00 0.00 +0.082 C ATOM 8 C UNL 1 -10.816 -2.990 0.656 0.00 0.00 +0.232 C ATOM 9 O UNL 1 -9.502 -2.805 1.229 0.00 0.00 -0.488 OA ATOM 10 C UNL 1 -8.810 -1.692 0.920 0.00 0.00 +0.162 A ATOM 11 C UNL 1 -7.447 -1.830 0.758 0.00 0.00 +0.054 A ENDROOT BRANCH 1 12 ATOM 12 C UNL 1 -5.154 -1.022 0.240 0.00 0.00 +0.139 C BRANCH 12 13 ATOM 13 N UNL 1 -4.292 0.168 0.332 0.00 0.00 -0.309 N ATOM 14 H UNL 1 -4.590 0.893 0.726 0.00 0.00 +0.149 HD ATOM 15 C UNL 1 -2.926 0.047 0.165 0.00 0.00 +0.215 A ATOM 16 N UNL 1 -2.222 -0.392 1.230 0.00 0.00 -0.222 NA ATOM 17 C UNL 1 -0.899 -0.464 1.024 0.00 0.00 +0.131 A ATOM 18 C UNL 1 -0.325 -0.124 -0.172 0.00 0.00 +0.054 A ATOM 19 C UNL 1 -1.136 0.298 -1.191 0.00 0.00 +0.131 A ATOM 20 BR UNL 1 1.534 -0.234 -0.411 0.00 0.00 -0.047 Br ATOM 21 N UNL 1 -2.467 0.400 -1.053 0.00 0.00 -0.222 NA ENDBRANCH 12 13 ENDBRANCH 1 12 TORSDOF 2 |
胺基嘧啶片段没对应的分支(BRANCH)没有出现在文件里,键N18_C17也没有出现在可旋转键列表里,这与ADT的prepare_ligand4.py准备的结果不同,可能会导致在分子对接过程中N18_C17键是刚性的而没有机会获得正确的结合模式。我建议尽量用ADT的prepare_ligand4.py来准备结构,或者用其它更准确的软件。
此外,电荷也是错误的:没有将删掉的氢上电荷合并到键连的重原子上,这将导致在采用力场打分错误(不影响VINA的docking,因为vina不使用用户给的电荷)。更多的PDBQT格式说明,请参见:What is the format of a PDBQT file?
进一步分析发现所有包含类似胍基片段的N-C键都被OpenBabel识别为不可旋转键,比如式2所示的常见2-氨基嘧啶片段。
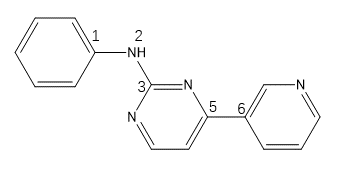
式2片段结构,具有三个柔性键
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 | (base) [gkxiao@master demo]$ cat S2.smi C1(NC2=NC(C3=CN=CC=C3)=CC=N2)=CC=CC=C1 S2 (base) [gkxiao@master demo]$ obabel -ismi S2.smi -opdbqt --gen3d --best REMARK Name = S2 REMARK 2 active torsions: REMARK status: ('A' for Active; 'I' for Inactive) REMARK 1 A between atoms: C_1 and N_2 REMARK 2 A between atoms: C_5 and C_6 REMARK x y z vdW Elec q Type REMARK _______ _______ _______ _____ _____ ______ ____ ROOT ATOM 1 N UNL 1 2.812 -0.545 0.128 0.00 0.00 +0.000 N ATOM 2 C UNL 1 3.296 -1.851 0.224 0.00 0.00 +0.000 A ATOM 3 N UNL 1 3.942 -2.233 1.334 0.00 0.00 +0.000 NA ATOM 4 C UNL 1 4.393 -3.501 1.427 0.00 0.00 +0.000 A ATOM 5 C UNL 1 4.180 -4.407 0.410 0.00 0.00 +0.000 A ATOM 6 C UNL 1 3.504 -3.932 -0.691 0.00 0.00 +0.000 A ATOM 7 N UNL 1 3.060 -2.669 -0.809 0.00 0.00 +0.000 NA ATOM 8 H UNL 1 2.532 -0.383 -0.830 0.00 0.00 +0.000 HD ENDROOT BRANCH 1 9 ATOM 9 C UNL 1 3.354 0.587 0.763 0.00 0.00 +0.000 A ATOM 10 C UNL 1 4.730 0.740 0.977 0.00 0.00 +0.000 A ATOM 11 C UNL 1 5.237 1.885 1.596 0.00 0.00 +0.000 A ATOM 12 C UNL 1 4.371 2.898 1.998 0.00 0.00 +0.000 A ATOM 13 C UNL 1 3.003 2.773 1.765 0.00 0.00 +0.000 A ATOM 14 C UNL 1 2.501 1.632 1.133 0.00 0.00 +0.000 A ENDBRANCH 1 9 BRANCH 4 15 ATOM 15 C UNL 1 5.110 -3.815 2.675 0.00 0.00 +0.000 A ATOM 16 C UNL 1 6.100 -4.795 2.757 0.00 0.00 +0.000 A ATOM 17 N UNL 1 6.791 -5.119 3.879 0.00 0.00 +0.000 NA ATOM 18 C UNL 1 6.496 -4.421 4.994 0.00 0.00 +0.000 A ATOM 19 C UNL 1 5.546 -3.417 5.038 0.00 0.00 +0.000 A ATOM 20 C UNL 1 4.853 -3.116 3.867 0.00 0.00 +0.000 A ENDBRANCH 4 15 TORSDOF 2 |
可以看到,键C1-N2与C5-C6是可旋转的,而键N2-C3被视为不可旋转:这要求初始的N2-C3一定是对的才有可能在后续的docking中得到正确的pose。OpenBabel的这个行为很危险,尤其是激酶抑制剂分子对接虚拟筛选时,比如PDB 1IEP的redocking计算(测试文件:1iep.zip),使用OpenBabel准备的配体文件(sti_ob.pdbqt)进行的Vina docking不能获得正确的pose,而使用adt准备的配体文件(sti_adt.pdbqt)却可以重现结合模式。
接下来开始准备vina的对接参数文件。
13. 通过共晶配体定义结合位点的中心
在第10步,我们将共晶配体提取出来,并保存为PDB格式。可以利用该配体来定义结合位点的中心坐标:
1 | obabel –ipdb ligand.pdb –obox |
其中输出如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | HEADER CORNERS OF BOX REMARK CENTER (X Y Z) 15.190 53.903 16.917 REMARK DIMENSIONS (X Y Z) 10.664 18.739 15.526 ATOM 1 DUA BOX 1 9.858 44.533 9.154 ATOM 2 DUA BOX 1 20.522 44.533 9.154 ATOM 3 DUA BOX 1 20.522 44.533 24.680 ATOM 4 DUA BOX 1 9.858 44.533 24.680 ATOM 5 DUA BOX 1 9.858 63.272 9.154 ATOM 6 DUA BOX 1 20.522 63.272 9.154 ATOM 7 DUA BOX 1 20.522 63.272 24.680 ATOM 8 DUA BOX 1 9.858 63.272 24.680 CONECT 1 2 4 5 CONECT 2 1 3 6 CONECT 3 2 4 7 CONECT 4 1 3 8 CONECT 5 1 6 8 CONECT 6 2 5 7 CONECT 7 3 6 8 CONECT 8 4 5 7 |
我们关注第2行,给出了配体的质心,我们利用该质心用于定义Vina的x,y,z中心坐标。
14. Vina的参数文件
Vina计算所需的参数可以通过键入vina命令来获得。参数可以从命令行输入,也可以从参数文件读入。假设我们的参数文件为config.txt,部分参数设置如下:
1 2 3 4 5 6 7 8 9 | receptor = protein.pdbqt #第11步准备的蛋白文件 center_x = 15.190 #第13步获得 center_y = 53.903 #第13步获得 center_z = 16.917 #第13步获得 size_x = 30.0 size_y = 30.0 size_z = 30.0 exhaustiveness = 8 cpu = 1 |
第7行exhaustiveness的值越大,AutoDock Vina搜索的越充分,计算所需的时间与之成线性增加;如果第8行不指定CPU的数量,则使用全部的cpu核心进行计算,所用CPU数越多,完成计算所需的挂钟时间越短。
15. 用VINA进行分子对接
现在我们用vina将第12步准备的配体对接到蛋白结合位点里,用纯命令行方式输入参数:
1 2 3 4 5 6 7 8 9 10 11 12 | vina --receptor protein.pdbqt \ --ligand ZINC000019632618.pdbqt \ --center_x 15.1 \ --center_y 53.9 \ --center_z 16.9 \ --size_x 30 \ --size_y 30 \ --size_z 30 \ --out vina.pdbqt \ --log vina.log \ --exhaustiveness 20 \ --num_modes 20 |
如果使用第14步准备好的参数文件,则命令可得到简化:
1 2 3 4 | vina --config config.txt \ --ligand ZINC000019632618.pdbqt \ --out vina.pdbqt \ --log vina.log |
其中–out为输出文件(pose预测结果);其中–log为日志文件。预测的pose可以直接重命名为pdb格式,用视图软件分析,比如pymol或Flare Viewer。其中Flare Viewer比起Pymol更加简单易用。
Figure 3. Flare分析STI与BCR-ABL的相互作用
避免误解的说明
本文采用PyMol进行初始的蛋白结构准备,仅是出于完成AutoDock Vina计算任务最基本的要求,并没有全面考虑蛋白结构准备的各个方面,比如:原子或残基缺失的修复、质子化状态与互变异构体的处理、侧链构象的选择、水分子的保留与否、金属离子的处理等等。正式的研究中应该充分考虑这些问题。PyMol并非蛋白结构准备的必须,也并非优选工具。有很多优秀的软件可以更可靠的进行结构准备。
OpenBabel用于将蛋白的PDB格式文件与配体的mol2格式文件转化为PDBQT格式文件非常方便,但是官方标配是AutoDockTools。并不了解OpenBabel与AutoDockTools两者之间的差异。读者需自行了解,但是OpenBabel无疑是非常方便的。
接下来可以做什么
- Virutalflow进行超高通量虚拟筛选
- 以预测的pose为起点用FEP计结合亲和力
- 以预测的pose为起点进一步用分子动力学模拟考察结合的稳定性
- 以预测的pose为起点用基于结构的方法生成药效团模型
文献
- Forli, S.; Huey, R.; Pique, M. E.; Sanner, M. F.; Goodsell, D. S.; Olson, A. J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11 (5), 905–919. https://doi.org/10.1038/nprot.2016.051.


