
基于结构与配体的方法协同研究RIPK1抑制剂的SAR
摘要:从探索化学空间到先导化合物优化,理解结构-活性关系(SAR)是药物发现诸多方面的基础工作。在本算例中,使用Forge的Activity Miner和Activity Atlas两个模块分析一系列苯并噁嗪酮类RIPK1抑制剂的SAR,演示了如何将Forge的SAR分析工具与Flare的蛋白质相互作用势及静电互补性分析协同工作,为配体设计提供了有用的见解。
作者:Sylvie Sciammetta. Investigating the SAR of RIPK1 inhibitors
编译:肖高铿/2021-03-31
前言
受体相互作用的丝氨酸/苏氨酸蛋白激酶1(RIPK1)是炎症和细胞死亡的关键介质。 它已成为有望治疗自身免疫疾病、炎性疾病以及癌症的靶标。 其独特的结构使得开发高选择性的小分子抑制剂成为可能。几个候选药物在临床上正取得进展,用于治疗诸如牛皮癣、类风湿性关节炎和溃疡性结肠炎之类的炎性疾病,诸如ALS和阿尔茨海默氏病之类的中枢神经系统疾病以及胰腺癌。
葛兰素史克(GSK)通过筛选DNA编码库发现了对RIPK1具有完全特异性的苯并噁嗪酮( benzoxazepinone)类RIPK1抑制剂[1]。 在本研究中,我们将使用Flare中的Activity Atlas和Activity Miner围绕苗头化合物GSK-481(图1A)探索苯并噁嗪酮类的SAR。
RIPK1抑制剂数据集的准备与叠合
数据集包含了活性范围从pIC50=4.9到10.3的46个化合物,可以从BindingDB下载[2]或者从Philip A. Harris等人[1]的文献支持信息中提取。与RIPK1结合的GSK-481的蛋白质-配体共晶结构是已知的,可从Protein Data Bank下载(PDB:5HX6)并在Flare中精心准备。
检查数据集分子结构的互变异体和质子化状态,并进行适当调整。然后使用标准的“Very accurate but slow”的构象搜索模式和子结构叠合允许的最大公共子结构叠合模式,将所有分子叠合到已公布的GSK-481的X射线晶体结构(PDB:5HX6)上,子 结构叠合时允许某些杂环旋转,并使用了“硬”蛋白排除体积。
在“Activity Atlas”中使用3D相似性要求对所有化合物进行叠合,并且对异常叠合和叠合噪声敏感。因此,建议对结合结果进行可视化检查以确保不存在任何异常,并且可用人工干预以改善次优叠合。因此,通过翻转/旋转噻唑环和苄基环来手动调节噻唑化合物1的取向。这以与整个数据集一致的方式对齐苄基环。得到的叠合结果如图1B所示。
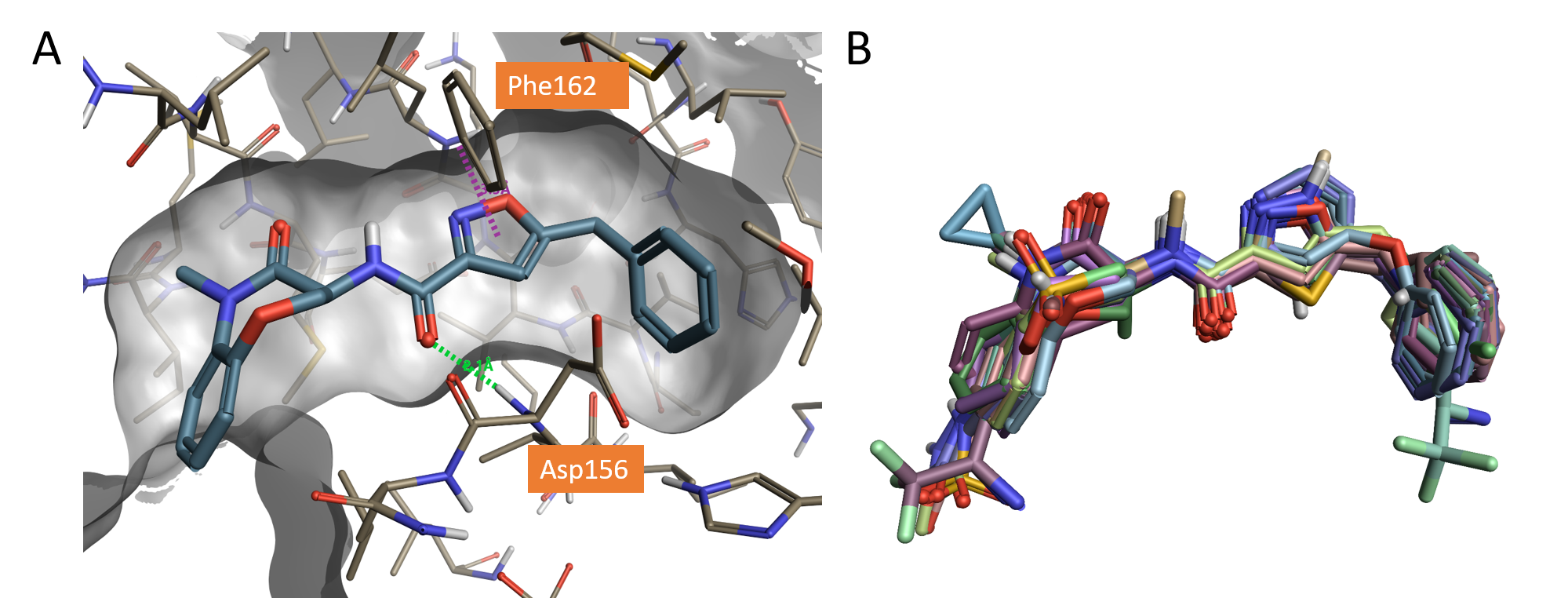
图1. A.共晶结构GSK-481(pIC50=8.8)结合到RIPK1的结合位点(PDB: 5HX6),绿色虚线为氢键相互作用,紫色虚线为pi-pi相互作用;B. 用Flare叠合到GSK-481(PBD:5HX6)的数据集化合物
用Activity Atlas进行SAR分析
Activity Atlas是分析一组叠合好的化合物的结构-活性关系随其静电、疏水和形状特性变化的概率方法。Activity Atlas特别适用于将大型数据表压缩成一张图片,将结构-活性数据总结成高度可视化的三维图,为新化合物的设计和优化提供信息。该方法使用贝叶斯方法定性地对数据进行全局观察,同时考虑分子正确叠合的概率。
Activity Atlas生成的活性悬崖(Activity cliff)汇总图提供了基于活性悬崖的SAR全景图。我们将重点分析使用“Normal”建摸条件下生成的SAR图,以了解RIPK1数据集的SAR特征。
形状与疏水性的活性悬崖总结
该数据集的形状活性悬崖概要图(图2A)表明,苯并噁嗪酮内酰胺上的小取代基可以增加对RIPK1的活性,但当存在较大取代基时,则活性降低。如图2A所示,在较大的不利区域内(品红色)封装了一个小的有利区域(绿色)。环醚部分的小变化是容许的,如该片段周围有利的(绿色)区域所示。几个不利区域(品红色)表明了该处存在一个紧密的结合点,其中仅允许微小的变化。

图2. 形状-RIPK1活性关系总结; B. 高活性化合物(pIC50=10.3-9); C. 底活性化合物(pIC50= 6.7-4.9)
对在蛋白质环境中叠合的配体进行初步的检查,似乎在蛋白质中几乎没有空间来容纳苯并噁嗪酮片段的构象小改变或结构变化。高活性分子(pIC50=0.3-9)显示出非常紧凑的叠合:GSK-481中只有苯并噁嗪酮氧被硫或NH取代是允许的,芳基苯并噁嗪酮环上的取代只允许在7位上进行(图2B)。相反,低活性分子(pIC50=6.7-4.9)的叠合表明与蛋白有许多的空间碰撞。
图3显示了苯并噁嗪酮片段内酰胺N-取代对活性的影响。与Activity Atlas完全一致,化合物2(pIC50 =7.49)中的NH用-NMe替换后(GSK-481,pIC50=8.8)提高了活性,更大的取代基如-NEt(化合物3,pIC50 =5.5)或N-cPR(化合物4,pIC50=5)会与蛋白相冲突,因此不可接受。
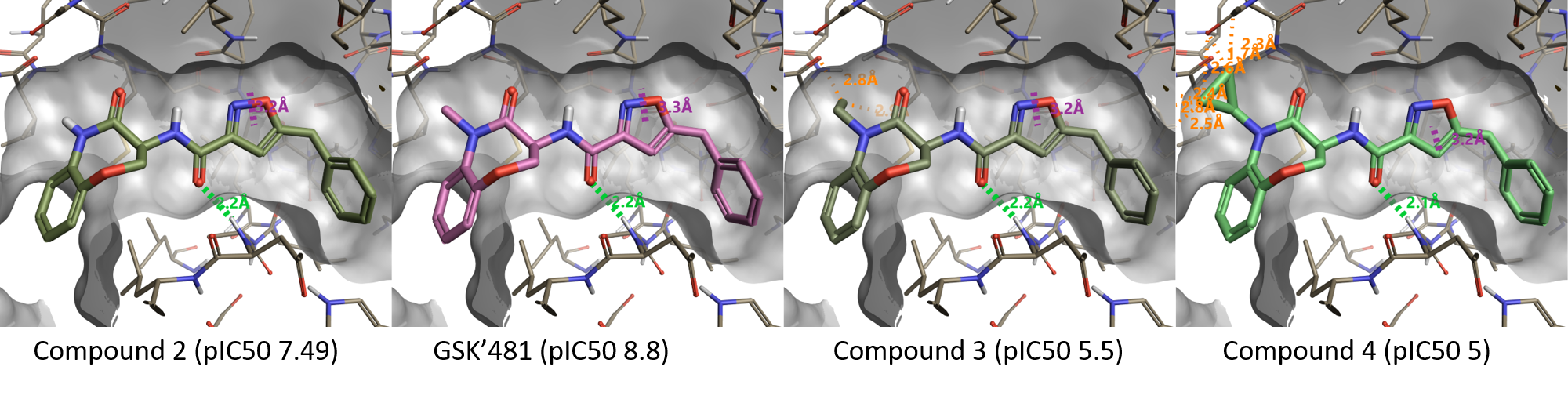
Figure 3. 内酰胺上比甲基大的取代是不允许的
对于该配体系列,疏水性活性悬崖分析显示出两个绿色区域,在这该区域引入疏水性取代基/片段对活性有利(图4A),在左侧有一个品红色的区域,在该区域引入疏水性片段对活性不利。 此外,我们在杂环(GSK-481异噁唑环)周围具有大面积的疏水不利区,这表明在该区域增加疏水性对活性有不利的影响。
对RIPK1的蛋白表面根据疏水性进行着色(图4B),异唑的CH和内酰胺NMe均指向亲水区域(蓝色),这与Activity Atlas等值图一致。
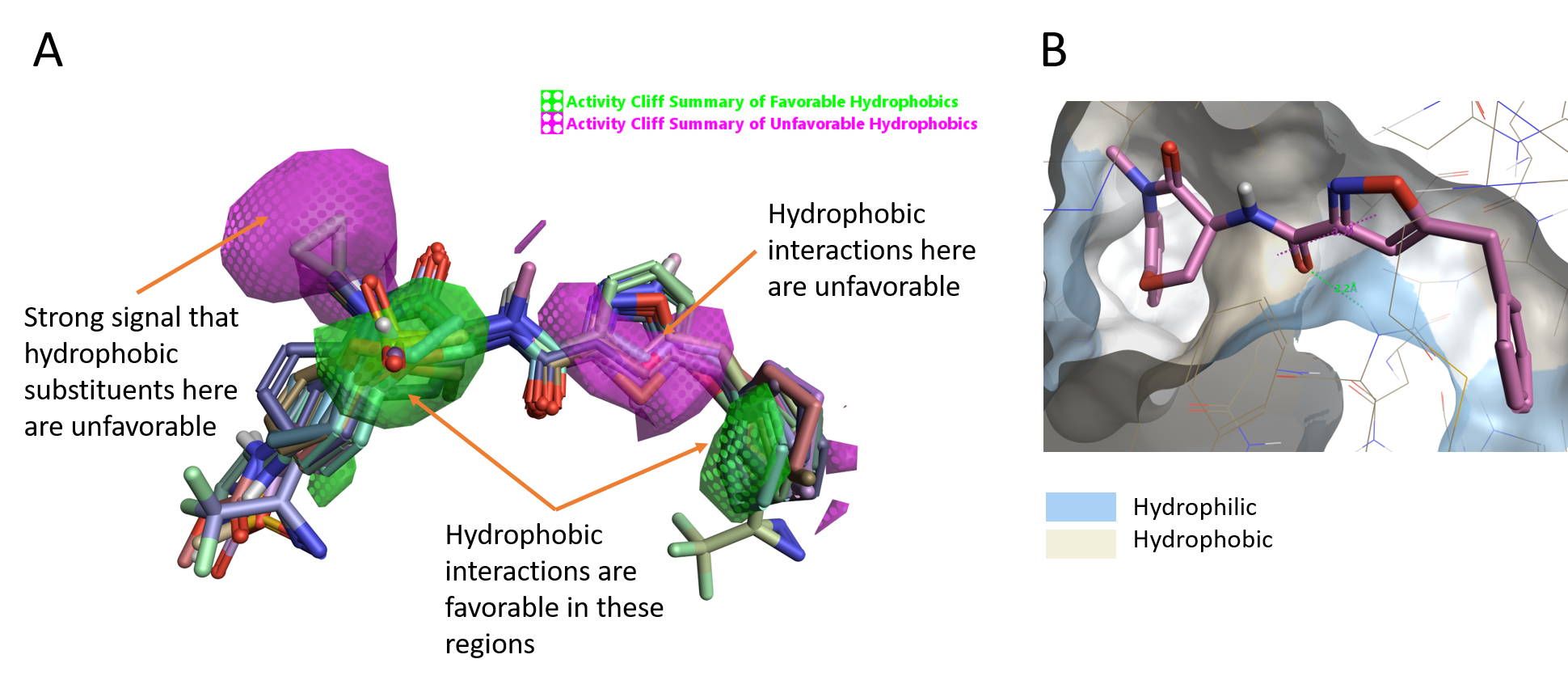
Figure 4. A. 疏水性活性悬崖分析; B. 用疏水性着色的RIPK1分子表面
静电势活性悬崖总结
静电势活性悬崖分析(图5)显示了负静电势(蓝色)与正静电势(红色)对RIPK1活性起关键作用的区域。

Figure 5. RIPK1的静电势活性悬崖分析.
在叠合好的配体的下方,增加负静电势对活性有利的区域显然与酰胺链接臂的羰基氢键形成有关系。在PDB 5HX6中羰基氧与Asp156骨架NH发生氢键相互作用(图1A)。
如图5所示,在叠合好的配体的链接臂上方的那个更大的正、负静电势有利区域与内酰胺羰基及5元环相关。然而,对RIPK1活性有利的效应不能简单地用经典的配体-蛋白相互作用来解释,借助蛋白相互作用势(Protein Interaction Potentials,PIPs)对RIPK1结合位点的静电分析可以帮我们进一步理解Activity Altas等值图报告的趋势。
PIP提供了蛋白结合位点详细的静电作用图,因此可以为理解配体结合、SAR和靶向蛋白的新分子设计提供宝贵的价值。可视化比较PDB 5HX6活性位点的PIP与噁唑5的配体场(pIC5=10.3,该系列中活性最强的化合物),发现噁唑右侧的正静电场正好与RIPK1活性位点的负PIPs对映。同时,配体的负静电场位于RIPK1活性位点正静电区域的中间(图6A-B)。恶唑5的静电互补图谱也证实了配体和蛋白静电之间的良好互补性(图6C)。
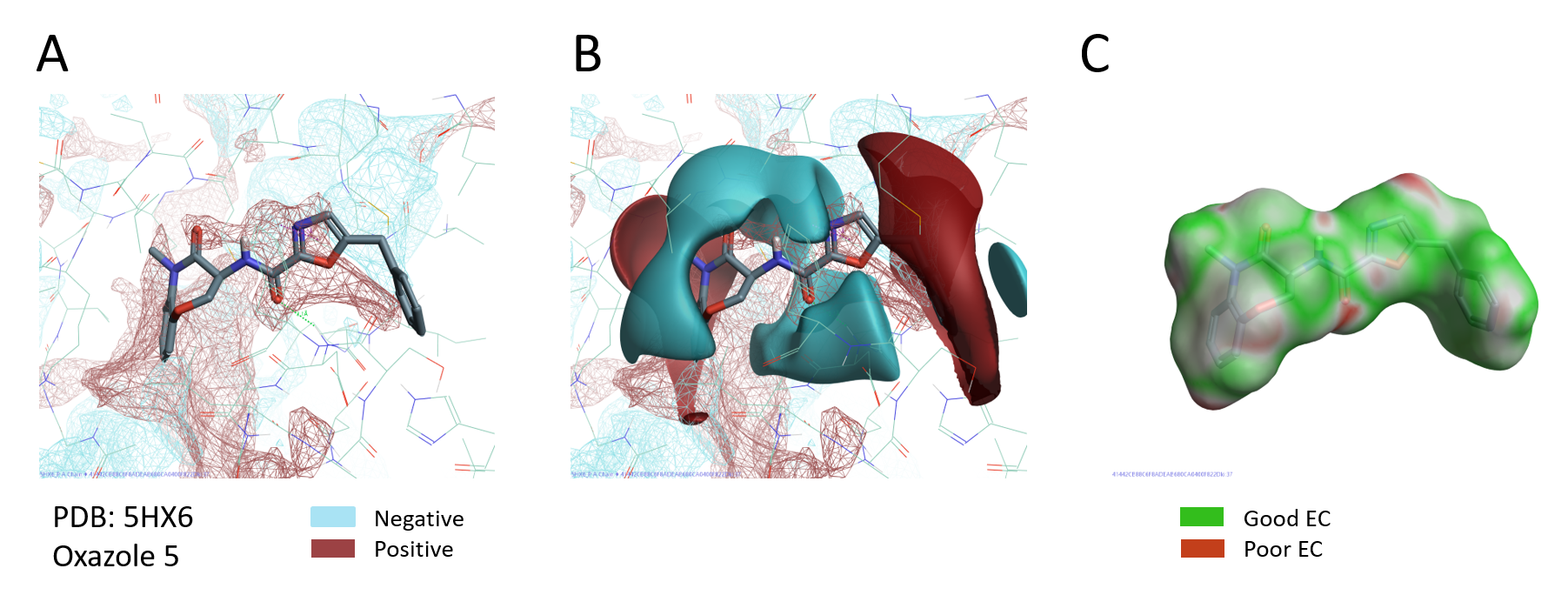
图6. A. RIPK1 (PDB 5HX6)的PIPs; B. 蛋白RIPK1的PIPs与配体噁唑5的静电场比较; C. 噁唑5与丹巴RIPK1之间的静电互补性
Activity Miner可以用来详细地分析配体静电场改变对RIPK1活性的影响。Activity Miner可以深入分析Activity Atlas的等值图以了解细微的分子间结构活性变化,并识别潜在的异常值。
用Activity Miner进行详细的SAR分析
Activity Miner能够让您在面对复杂的SAR时能够游刃有余,它可以突显出分子对之间的关键活性变化,并为活性变化给出清晰的根本原因。它还为如何利用这些知识进行未来的设计迭代提供了灵感。Activity Miner使用视差(disparity)或活性悬崖(activity cliffs’)的概念来突显在SAR全景中结构的小变化会导致活性的大变化的区域。
Activity Miner与配体场差异和PIP结合使用,可以用来研究SAR的关键区域。我们对含有17个分子的子集进行了研究,这些分子由GSK-481的异恶唑环片段用表1的各种杂环替换而来。
Table 1.不同的环取代及其pIC50
Activity Miner的Top pairs表列出了选定活性的最高落差分子对,这让你能够看到对PIRK1 PIC50影响最大的分子结构变化。探索最靠前的几个分子对的配体场差异(一个分子中的配体场比另一个分子中的配体场更正或更负的区域)(图7和图8)可以证实:杂环左侧场越负(通常与芳香氮有关)、杂环右侧场越正则对PIRK1的pIC50越高。这与Activity Atlas等值图以及RIPK1的PIPs分析相一致。

图7. Activity Miner的“Top Pairs”表列出了活性落差最高的分子对。一个分子比另一个分子越正(红色)或越负(蓝色)的区域为配体场差异,配体的静电势图为场差异(field difference)。每个top pair,活性最强的分子展示在左边、活性差的那个分子展示在右边。同时也展示了蛋白RIPK1的PIPs:红色为正静电势,蓝色为负静电势。

图8. Activity Miner “Top Pair”表的其它几个例子。配体静电以场差异的方式呈现于RIPK1结合位点PIP的上方:红色为正静电势,蓝色为负静电势。
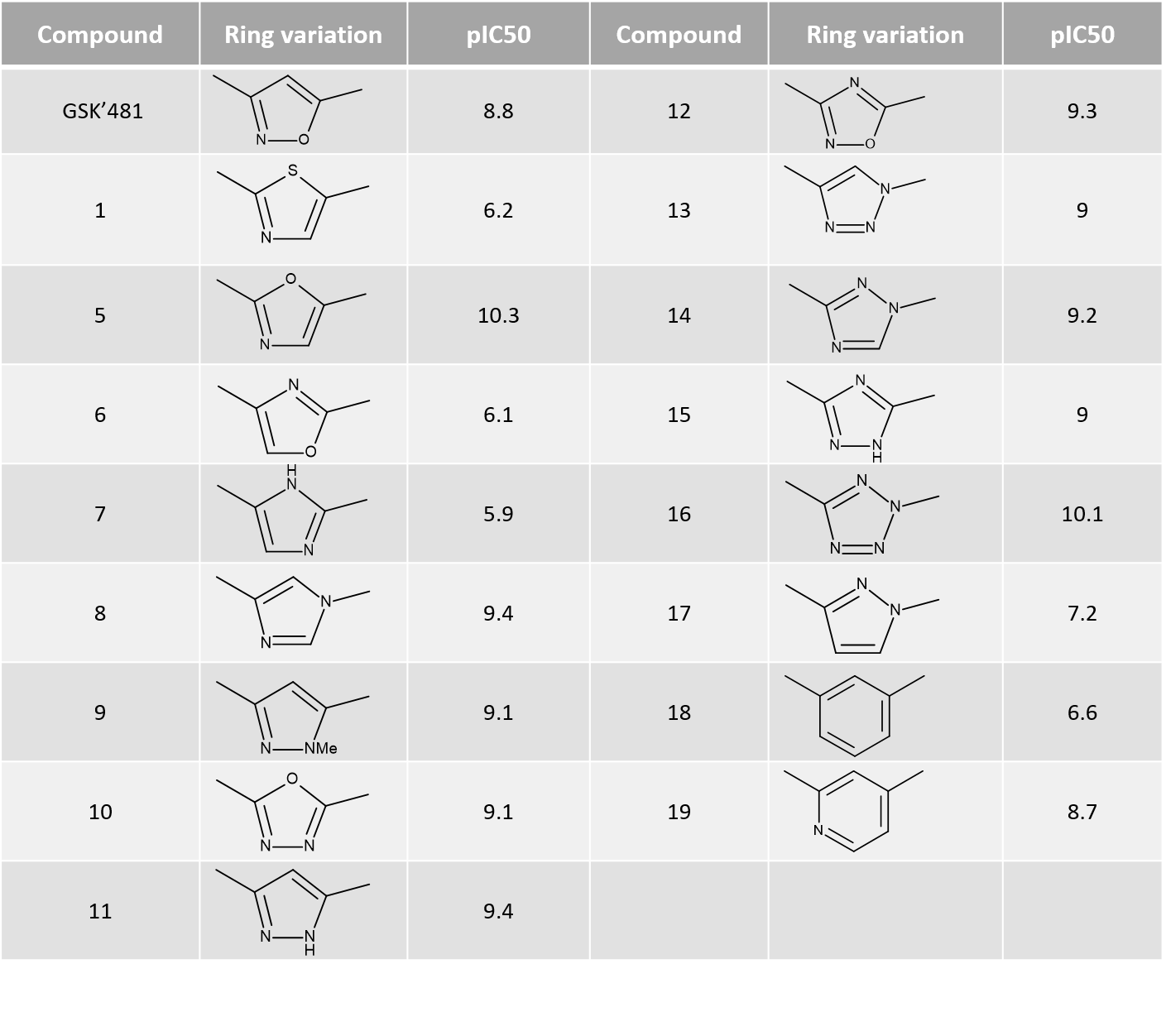
使用Activity Miner对SAR进行的深入研究还可以从总体趋势发现可能的例外(异常值)。噻唑1(pIC50=6.2)具有与噻唑5(pIC50=10.3)有着非常相似的静电特性,但活性要小得多(图9A)。一种可能的解释是,较大尺寸的硫原子可以改变噻唑和/或苄基的取向,导致其以次优选的构象结合于蛋白口袋。噻唑的高亲脂性也可能导致化合物1的活性较低,因为噻唑环位于蛋白中亲水区域附近(图9B-C)。
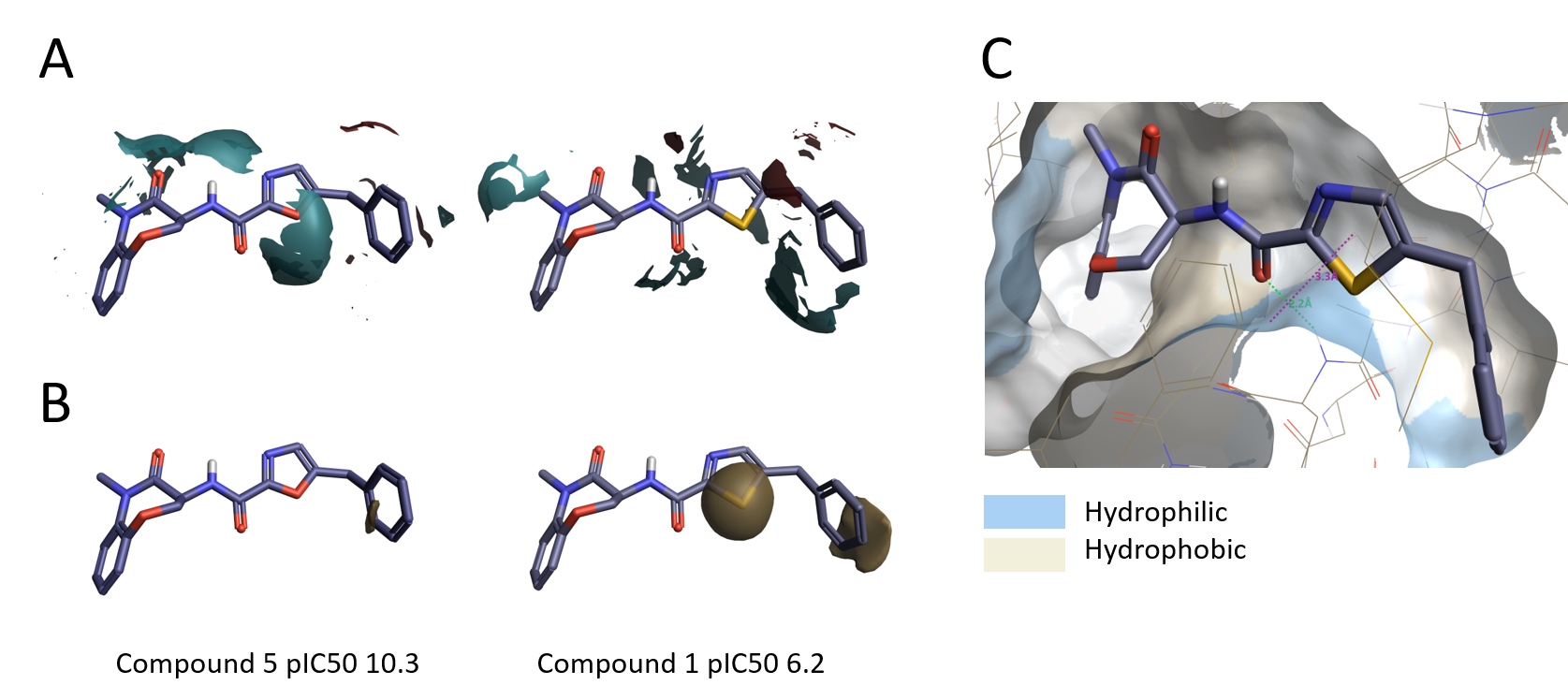
图9. A. 噁唑5与噻唑1之间的配体静电场差势图;B.噁唑5与噻唑1之间的配体疏水场差势图;C.噻唑1在RIPK1结合位点里,蛋白表面用疏水性着色。
用静电互补性分析SAR
通过比较配体和蛋白在配体溶剂排除表面的所有顶点处的静电势值可以计算配体对蛋白质的静电互补性(Electrostatic Complementarity,EC)[3]。与蛋白静电互补的配体表面区域被着色为绿色,而有静电冲突的区域被着色为红色。
为了适用于不同蛋白-配体场景,EC打分采用三种不同指标来量化配体-蛋白的静电互补性。图10显示了表1中一些化合物的EC图,以从左到右按RIPK1活性降序显示。

Figure 10. 围绕异噁唑环进行结构改变后的静电互补性分析。绿色:良好的静电互补性;红色:静电冲突.
可以观察到明显的定性趋势: 随着配体活性的降低,出现更大的静电冲突红色区域。图11A所示的RIPK1 pIC50-Complementarity ρ打分图证实了这一趋势。两个明显的异常是噻唑1与6。如前所述,由于硫原子的大小和亲水性,噻唑1可能具有稍微不同的结合模式,噻唑6的叠合构象显示出内部的氢碰撞与孤对电子排斥(图11B)。当结合到RIPK1的活性位点时,这两个化合物极可能采取了一种完全不同的构象。
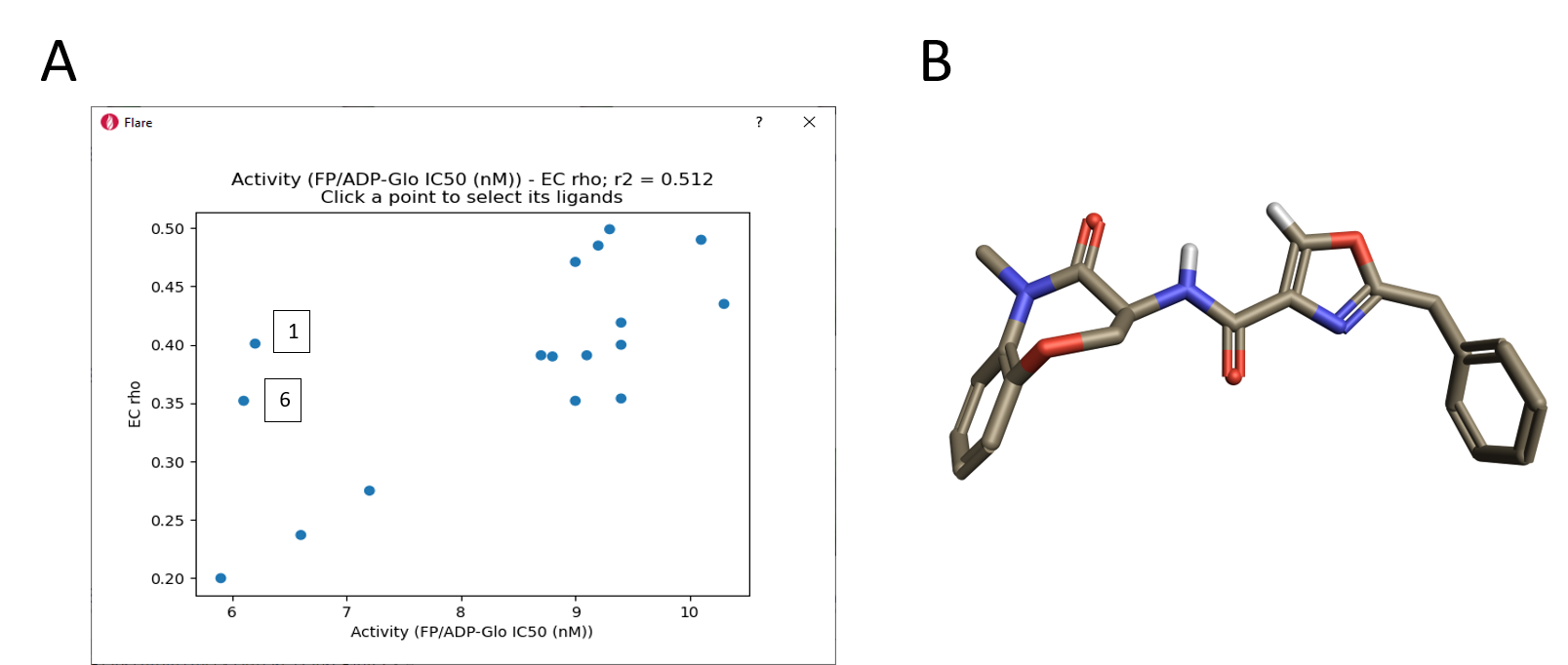
图11. A. EC打分与活性的相关性;B. Figure 11. A. Correlation of EC scores with activity range; B. 噁唑6的叠合构象
结论
将Forge方法整合到Flare的完全集成方法(定于2021年夏季发布)可实现基于配体与基于结构方法之间的协同相互作用,从而为SAR分析、配体设计和药物发现提供了更有效的方法。
文献
- Harris, P. A.; Berger, S. B.; Jeong, J. U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C. A.; Cox, J. A.; Dare, L.; Dong, X.; Eidam, P. M.; Finger, J. N.; Hoffman, S. J.; Kang, J.; Kasparcova, V.; King, B. W.; Lehr, R.; Lan, Y.; Leister, L. K.; Lich, J. D.; MacDonald, T. T.; Miller, N. A.; Ouellette, M. T.; Pao, C. S.; Rahman, A.; Reilly, M. A.; Rendina, A. R.; Rivera, E. J.; Schaeffer, M. C.; Sehon, C. A.; Singhaus, R. R.; Sun, H. H.; Swift, B. A.; Totoritis, R. D.; Vossenkämper, A.; Ward, P.; Wisnoski, D. D.; Zhang, D.; Marquis, R. W.; Gough, P. J.; Bertin, J. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60 (4), 1247–1261. https://doi.org/10.1021/acs.jmedchem.6b01751.
- BindingDB.Retrieved from https://www.bindingdb.org/entry/50049028. BindingDB DOI:10.7270/Q2SQ92NR
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. J. Med. Chem. 2019, 62 (6), 3036–3050. https://doi.org/10.1021/acs.jmedchem.8b01925.


