
摘要:本文介绍了最新发布Flare V5中Flare FEP的特性介绍,包括:不影响准确度的情况下近乎1倍的速度提升;自动的中间体添加;改进的拓扑构造容纳更多的分子;提升的预测性能与准确度;易于使用、易于结果分析。
原文:Jenny Brookes(27 July 2021). Retrieved from https://www.cresset-group.com/about/news/flare-fep-speed
编译:肖高铿
自由能微扰 (FEP) 计算为药物猎人提供了高精度的配体结合亲和力预测工具。根据实验值评估,该方法准确度可优于1 kcal/mol,因此可以用于选择最有潜力的分子以便对之进一步开发与合成。然而,使用 FEP的常见障碍通常是计算非常复杂,即难以设置和执行,并且还可能需要花费很长时间才能完成计算。因此,就用户和总计算时间而言,FEP以其昂贵而闻名。Flare FEP作为基于配体与基于结构设计平台Flare软件的一个组成部分,它显著地降低了这一障碍,因为可以快速、轻松地得到非常准确的FEP结果。 下面我将描述一下研究人员可以期待从Flare FEP中获得什么样的速度、精度和易用性,并可提供测试版以便您亲自对软件进行评估。
巨大的速度提升、更快地看到FEP结果
最近发布的Flare V5显著提高了Flare FEP的计算速度。对于中型体系,Flare V5计算至少比Flare V4快2.5-3.5 倍。以Wang等人[1]的基准测试集中的“TYK2”为例(包括 11 个配体、14 个链接和28个微扰),该算例用Flare V5需要使用106个GPU小时,用Flare V4需要470个GPU小时。如果您有一个10张GPU的集群(Flare FEP 计算需要OpenCL或NVIDIA CUDA的GPU支持),相当于11个分子大约需要10.6小时,而相比之下Flare V4则需要47小时,这大大改善了看到FEP结果所需的时间。
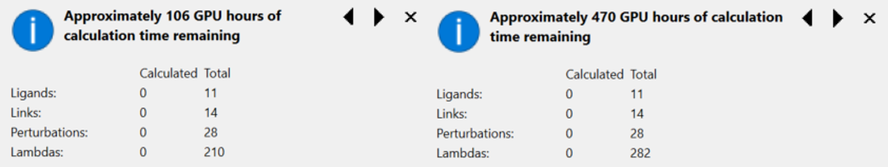
图1. Flare FEP计算的贴士弹出窗口。用TYK2算例来比较Flare V5(左)与V4(右)的Flare FEP计算差异。配体的数量、链接数、微扰的数量都一模一样,但是Lambda窗口数量有差异,这使得Flare V5比V4的计算速度加速了4倍。
Flare FEP快速的计算是下面讨论的多种优化的综合结果,其中一个最显著的算例在Lambda设定上有了非常大的提升,
自适应Lambda规划提升30%的速度
扰动之间的λ窗口表示配体从A转换到B时所进行的增量跳跃。手动设置Lambda窗口可并非显而易见但往往很耗时,而且当某些扰动比其他扰动更棘手时,设置一个全面的默认值是很浪费时间的。
如果您自己决定亲自手动设定每个链接的Lambda 窗口(可能有数百个),您无法在计算之前知道您是否高估或低估了Lambda窗口。如果采样不足并且Lambda之间没有足够的重叠,则需要放弃结果并重新计算(图 2 – 左)。如果使用超过所需的Lambda窗口而过度采样,则会以更昂贵的计算为代价达到足够的重叠(图 2 – 中间)。除非你的选择完全正确,否则任何一种方式都会浪费计算时间。
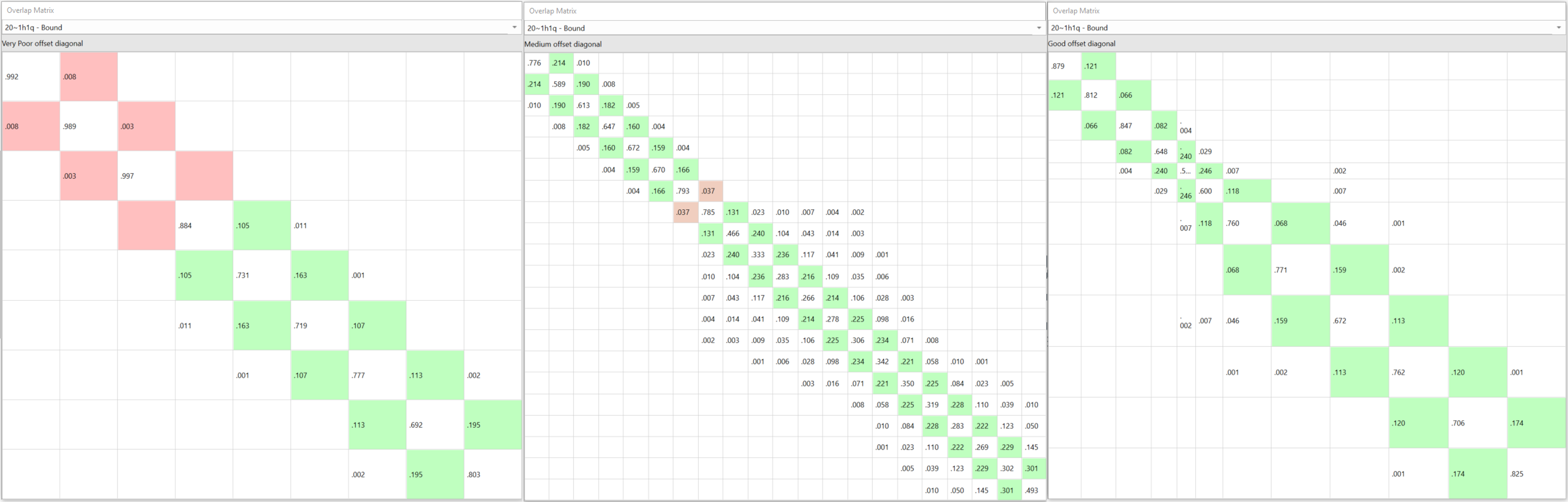
图2. 基准数据CDK2(PDB 1h1q)连接数在20左右时的采样不足(左:9 个 Lambda 窗口)、过度采样(中:21 Lambda窗口)与自适应Lambda规划(右:12 Lambda窗口)的比较。
如图2(右)所示,这些问题通过新的“自适应Lambda规划”来消除,在FEP计算开始时,快速(50ps)的预计算用来确定每个特定扰动所需的Lambda窗口最佳数量。
表1. Flare V5与V4中所需的总Lambda窗口数以及减少百分比。*假设不需要使用更多Lambda窗口而重新对链接计算。
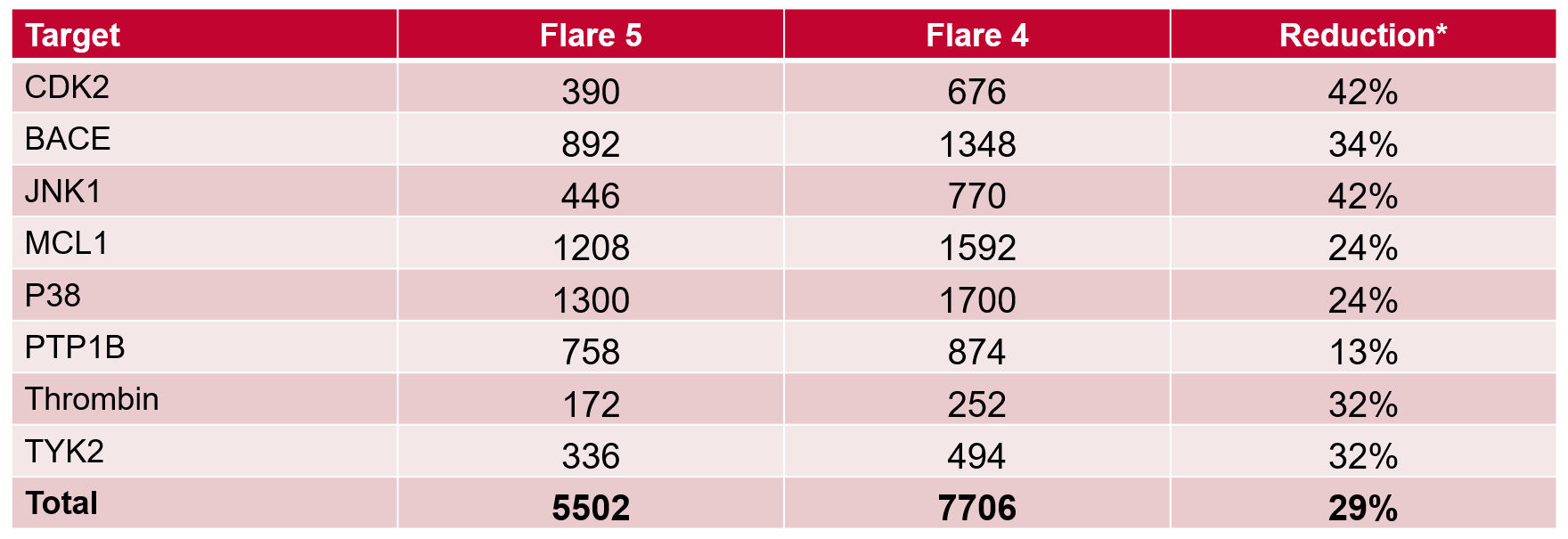
对于Wang基准测试中的8个数据集,使用Flare V5中的自适应Lambda 采样所需的总Lambda窗口要少得多,而在Flare V4中为每个链接选择一组固定的9个Lambda窗口(表 1)。事实上,计算量减少了约 30% 是低估的,因为它没有计算任何由于采样不足而被丢弃的计算(图 2-左),或者由于过度采样而比完成计算所需花费的超出的部分 (图 2–中)。
改进的积分器:具有更快的速度
我们优化了Flare FEP分子动力学模拟计算使用的积分器。新的Langevin Middle Integrator更稳定,允许使用更长的时间步长。通过添加少量的氢质量再分配(Hydrogen Mass Repartitioning,HMR),其中氢质量根据它所附着的重原子进行再分配,而不改变总质量,我们已经能够将默认时间步长从2fs增加到4fs,而不损失稳定性。这使得每个链接所需的GPU时间减少了近两倍。
使用截短的八面体水盒子:更少的水分子意味着更短的模拟时间
截短的八面体水盒子是在 MD 阶段实施的另一个开发,它减少了 Flare FEP的计算时间。八面体形状对MD来说是更效的模拟单元,因为它更接近于球体。这减少了模拟中所需的水分子数量,从而减少了模拟的长度。对于一个小的示例蛋白,使用八面体盒子作为模拟单元,与使用立方体盒子相比,原子数从17.5K 减少到10K 原子(图 3)。测试还表明,Flare V5中较小的6Å水盒子缓冲(相对于Flare V4中的10Å)足以模拟溶液中的蛋白-配体复合物(与附近单元之间没有相互作用)。
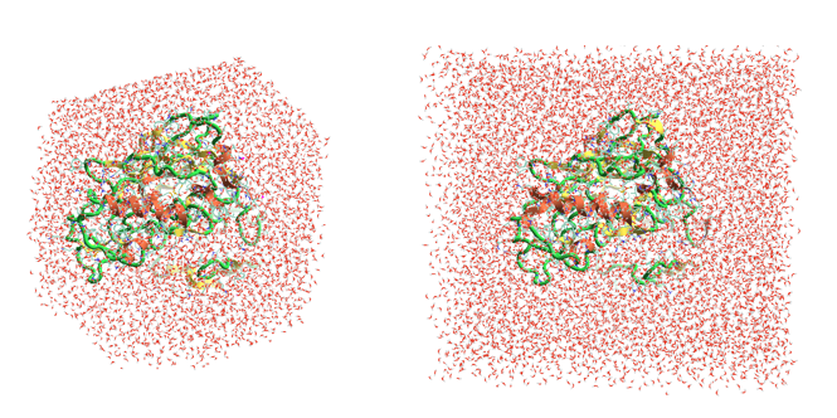
图3. Flare V5使用新的截短的八面体水盒子(图左)含10K个原子, 相比Flare V4使用的立方体水盒子(图右)的17.5K个原子,减少了MD模拟中水分子数。
优化的工作流与可伸缩性
在Flare V5中,我们在后台进行了许多改进,以允许在更大的数据集上运行FEP计算。如果您有许多分子,那么设置微扰网络的计算(即选择哪些分子对来进行微扰计算)可能需要大量时间。新代码不仅使这类计算速度更快,而且还支持跨多个CPU核心以及多节点的并行化计算。因此,可以轻松为约150个配体的数据集生成标准微扰图(拓扑构造),并且可以为500多个配体设置星形图(图 4)。
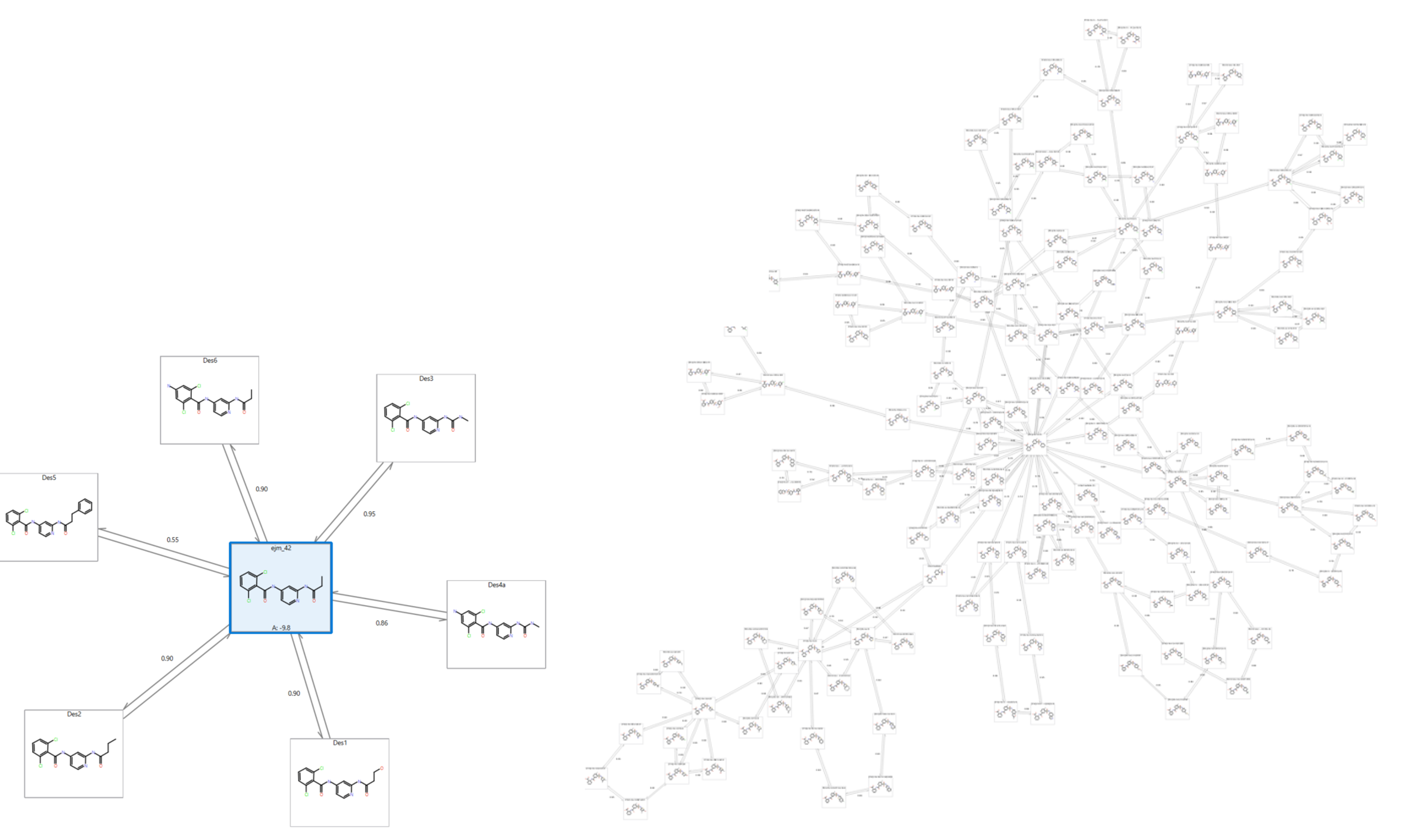
图4. 含150个配体的星形图(左)与标准微扰图(右)比较。星形图可以容纳500个配体,本文出于清晰的示例原因,仅展示了少量化合物。
在这个尺度上工作可以让你快速专注于值得追求的分子。例如,对SPARK生成的500个结果以快速模式、星形拓扑构造进行Flare FEP计算实验,然后进一步使用双向链接、正常图拓扑构造对最有希望的结果(例如从原始500中取出最佳的50个)进行精细的计算。这使您能够快速地筛选最佳分子,并最终通过稳健和准确的结合自由能预测来进行优先性排序,创建小得多的分子子集用于合成与活性测试。
提升了预测性能与准确度
最重要的是,Flare V5速度的提升并不影响Flare FEP的预测性能和准确性。Flare V5 仍然提供可靠的结果,正如之前《炼金术法自由能计算评估结合亲和力》[2]一文所验证的那样。Flare FEP计算的平均无符号误差(MUE)值与文献报道的同类方法的MIUE值相比非常有利,除了一个例子外,所有的MUE都小于1 kcal/mol(图5)。
![图5.使用Wang等人<sup>1</sup>的基准数据集,比较Kuhn等人<sup>[2]</sup>报道的MUE(蓝色)与Flare V5计算的MUE(黄色)](http://blog.molcalx.com.cn/wp-content/uploads/2021/08/2021081813594853.png)
图5.使用Wang等人[1]的基准数据集,比较Kuhn等人[2]报道的MUE(蓝色)与Flare V5计算的MUE(黄色)
提高了准确性,特别是对于较奇特或参数化较差的分子
自定义力场向导使您能够轻松选择感兴趣的扭转角并编辑分子以减少为获得良好扭转参数的非必要原子。各种量子力学方法和基组的下拉列表使您能够创建定制的参数以补充 AMBER GAFF 或 GAFF2。
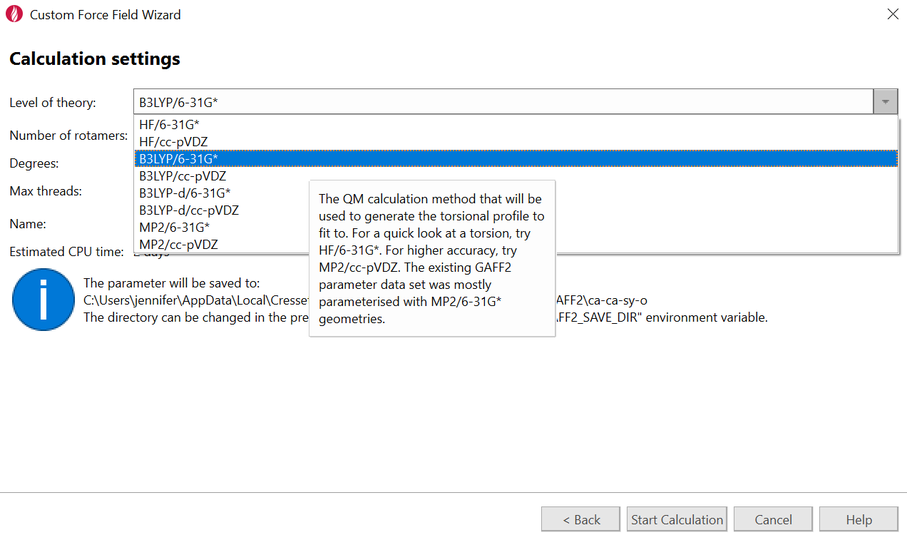
图6. 自定义力场参数向导提供了多种QM方法与基组,为您的数据集添加自定义的GAFF与GAFF2力场参数。
更高效的FEP方法使得Flare FEP更易于使用
除了Flare FEP在运行时间上的巨大改进外,还有许多的功能增强,使得FEP模拟的设置过程更简单、更高效。这些发展在微扰网络图的生成时是可以体会到的。
中间体自动化
微扰之间的链接打分值(图 7)告诉您配体 A 转换为 B 的难易程度。打分值为 0(非常不同)到 1(完全相同)范围内的一个值。
对于某些配体,打分值非常低(例如小于0.4)的链接不会产生好的结果。在这种情况下,建议添加“中间体”结构以促进 A⇒B 过渡并改善您的结果。 手动添加中间体结构很麻烦:您必须设计、编辑您的分子并将其叠合到原始数据集上,您可能需要多次执行此操作,具体取决于配体数据集的大小和细节。这个过程的自动化极大地提高了可用性:Cresset新的基于规则的启发式算法在需要的地方创建中间体,并且很好地改进了Flare FEP模拟。
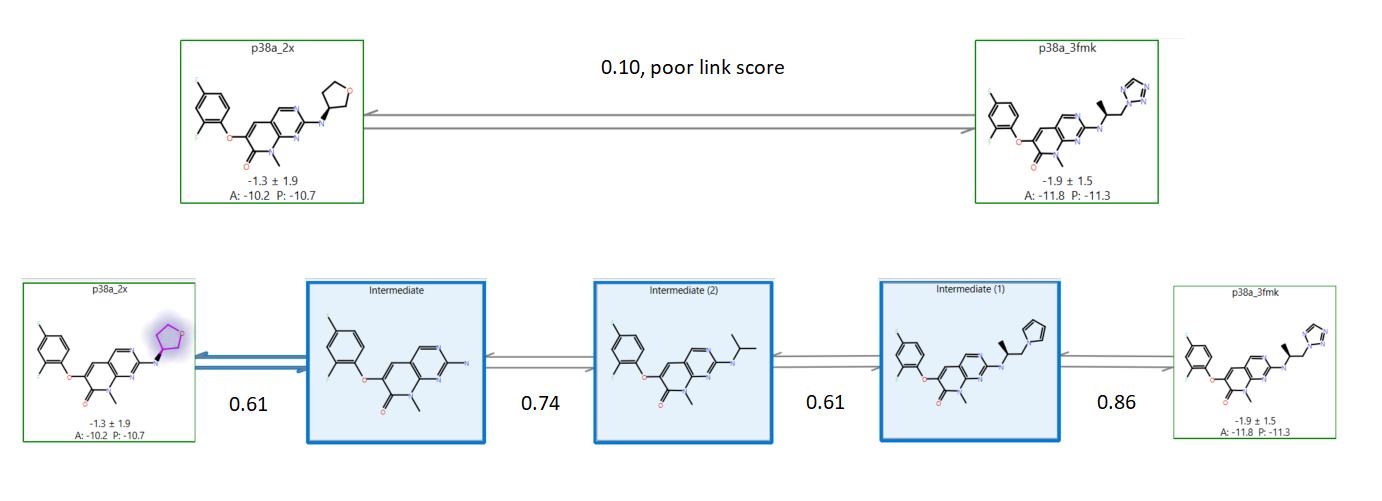
图7. 中间体生成过程的演示。在没有中间体的时候,绿色盒子里的两个已知分子间的链接打分值很差(0.10),A与B非常不相似。增加了中间体(蓝色盒子)直到链接打分优于0.6极大的改善了计算结果的质量,而无需用户任何干预。
易于分析的结果
使用“开箱即用”的 Flare FEP(图 5),通常可以提供快速且非常好的初始结果。一旦您仔细准备并设置了蛋白靶标,只需几分钟即可生成初始微扰网络并开始Flare FEP计算。
对微扰图预计算进行完整性检查,并尽可能进行计算后的优化,可以进一步提高结果的准确性。
用户友好的Flare FEP界面有助于检查并进一步细化初始结果。它使所有重要检查项目都可见:平衡配体的三维结构,重叠矩阵的质量,循环闭环误差、日志中任何潜在问题的链接,以及活性绘图等等。
1 分钟视频《Flare FEP结果的轻松分析》展示了Flare FEP中结果的分析和优化的便利性。请注意,带有统计数据的活动图,其数据点与配体之间是同步的,您可以在3D视窗中可视化这些数据点与配体结构,以检查蛋白和水的相互作用。在本算例中,如果您发现存在“较差的叠合”,您可以在“叠合矩阵”选项卡中进行检查,看看是否有可以通过快速修改链接来改进的窗口。
您还可以手动调整和删除中间体结构,如果你认为它们是不必要的。 然而,一般来说,与一次进行大的转换步骤相比,该算法提供了适当数量的分子来提高精度。现在还可以选择在不清除任何完整连接的情况下强制配体重新平衡,并将平衡后的复合物推回到Flare进行3D分析。所有这些发展都有助于简化故障排除和改进FEP结果,以提供您有信心的预测。
文献
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M. K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137 (7), 2695–2703. https://doi.org/10.1021/ja512751q.
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A. S. J. S.; Mackey, M.; Michel, J. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60 (6), 3120–3130. https://doi.org/10.1021/acs.jcim.0c00165.


