
摘要:本文分享了Cresset药物发现咨询服务团队一个真实项目的描述,在这个案例中,尽管遇到很多困难,但从专利突破发展到结构新颖、并具有选择性的系列化合物发现、并最终解决了与靶标有关的许多问题,进展相对较快。该项目已步入正轨,Cresset的建模提供了独特而有价值的见解,随着项目的推进,在今后的应用中继续为项目增值。
作者:Martin Slater. Cresset, New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK
编译:肖高铿/2023-04-12
前言
药物的研究和开发从来不是一件容易的事。真正的研究总是牵扯各种起起落落、错误的假设、糟糕的数据和不明确的目标。成功之路往往是曲折的、布满障碍和陷阱,充其量是一个相当混乱的过程。
本文是对一个真实项目的描述,包括了所有的缺陷。无论是在学术界还是在制药工业界进行的项目中,这都是很典型的。最终,随着从生物学、化学和建模中获得了新的数据,如果在此过程中仔细应用,则会取得进展并获得知识。
在这个案例中,尽管有下述困难,但该项目从专利突破发展到结构新颖、并具有选择性的系列化合物发现、并最终解决了与靶标有关的许多问题,进展相对较快。该项目已步入正轨,Cresset的建模提供了独特而有价值的见解,随着项目的推进,在今后的应用中继续为项目增值。
概述
从2012年到2014年,Cresset咨询服务团队与Newcastle大学和Sygnature Discovery就MRC资助的骨关节炎项目开展了三方合作,为计算建模工作做出了贡献。Strathclyde大学1和Newcastle大学2进行了重要的实验,确定了蛋白Matriptase是导致关节中胶原降解疾病通路的关键介质。该蛋白靶标3是丝氨酸蛋白酶家族的成员,Steinmetzer4改造了尿激酶拟肽抑制剂并获得该靶标的共晶结构。这些抑制剂含有众所周知与P1口袋相互作用的苯甲脒基,该片段对这类蛋白酶的活性至关重要。
建模工作的目的是为Newcastle大学提供新的Matriptase抑制剂先导化合物,不但要求具有自主知识产权,而且更重要的是具有更有利的体内活性特征。
方法
模板准备(Templating)
对Matriptase晶体结构(PDB 2GV6、2GV7和3P8G)的分析可得到Steinmetzer SAR系列化合物的初步结合假设。然而,该系列中活性最好的化合物没有共晶结构,因此,首先对Cresset专有的XED力场模拟共晶结构的能力进行验证。这可用2GV6配体本身进行尝试。这个实验得到令人惊讶的结果(图1,品红色结构),并促使我们对该体系进行更详细的研究,包括其他靶标和结构,例如尿激酶(PDB 2VNT)和胰蛋白酶(PDB 1K1L)。
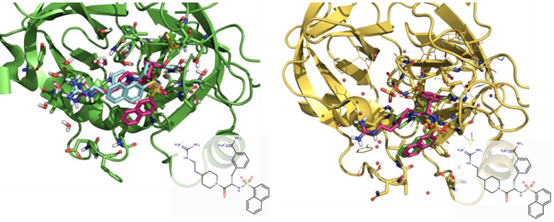
图1. 左:Matriptase二聚体(PDB 2GV6)显示为绿色飘带(仅显示其中一个单体),其磺酰胺抑制剂显示青色,模拟的单体形式的抑制剂显示为品红色。右:胰蛋白酶(PDB 1K1L)显示为黄色飘带,其抑制剂显示为绿色,叠合上去的模拟的Matriptase配体显示为品红色。
进一步的分析显示,在晶体学中观察到的蛋白质-配体复合物实际上是二聚体(青色结构),涉及到重要的相互作用,包括蛋白质对蛋白质和配体对配体,这决定了最终观察到的结合模式。这一事实解释了为什么这些配体的一些极其疏水的部分被认为是溶剂暴露的难题(也就是说,它们没有暴露于溶剂),但也强调了为了正确解释SAR,必须考虑靶向单体和二聚体蛋白混合在一起的可能性。
令人满意的是,XED力场非常精确地模拟了在单体形式蛋白质中能量最小化的抑制剂,实际上也是基于Steinmetzer SAR最佳配体候选之一(图2)更可能的方案。
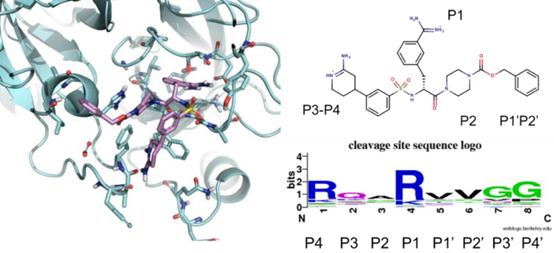
图2. 左:Matriptase与磺胺类抑制剂的结合模型(PDB 2GV7),Matriptase以青色飘带显示,单体形式的磺胺类抑制剂以淡紫色模型显示。右:抑制剂的组成部分划分与Matriptase丝氨酸蛋白酶子口袋的结合归属以及Matriptase酶的底物残基偏好示意图。
Blaze虚拟筛选
掌握了活性最强、但碱度最小配体的结合假设之后,就可以用Cresset的虚拟筛选技术Blaze从300多万个有商品可供购买的化合物库中寻找替代骨架。由于参比化合物具有一个苯甲咪片段,主要倾向是高TPSA(大于145)和高碱度(pKa=11.6)等缺陷,所以我们要确保在搜索结果中删除胍类和苯甲咪类化合物。这个决定不是轻易做出的,因为苯甲咪是决定药效的关键因素。然而,如果愿意从一个潜在的薄弱起点进行优化,这将为最终实现口服生物利用度的目标提供最佳机会。因为配体相当大,所以我们的虚拟筛选策略是将模板分成三个适当大小、更具类先导的分子,如图3a和3b所示。
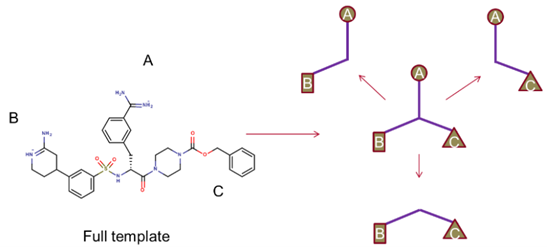
图3a. Blaze虚拟筛选模板分子的碎片化策略
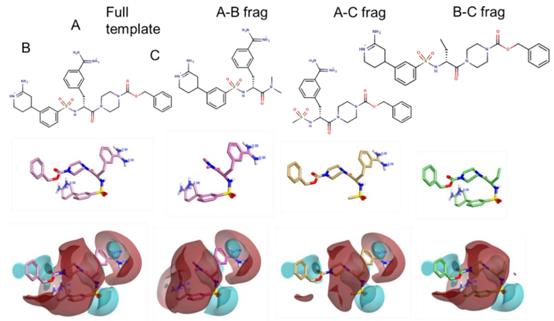
图3b. 参比模板碎片化为3个类先导分子及其相应的静电场表面
不幸的是,最终仅购买并筛选了一小部分虚拟筛选结果。在142个分子中,仅化合物0154对Matriptase有明显的活性,尽管活性相对较弱(IC50 = 30 μM)。
该分子是从A-B片段发现的,因此缺少模板分子的C端。该苗头化合物不具高碱度,因此从其优化潜力来看令人感兴趣,它满足了我们所期望的性质特征。然而,在第二轮对可供商品销售的类似物测试结果让人失望,在29个化合物中没有发现任何活性更强的分子。
平行专利突破
与此同时,化学合作伙伴在最近报道的合成抑制剂专利突破策略上也同时在努力。这种方法的缺点是,该专利突破来源于一个对称的三碱基分子,因此优化,特别是ADMET特性的优化,将是一个问题。此外,由于对称性的起始配体固有的模糊性,使得结合假设的建模变得更加复杂。
然而,合成努力获得了一些与0154活性相当的化合物(图4),可以通过更便捷的化学方法获得,并具有独立自主的知识产权。

图4. 29个Blaze苗头化合物的类似物和早期专利突破例子的活性,表达为在10μM浓度下对Matriptase的百分抑制率。
经过系统的化学优化,具有合理活性的化合物开始出现。在对结合模式假设进行了至少一次修改后,对项目 “按需 “建模的支持终于开始从新系列的SAR模式中揭示出一些关键的结合相互作用。
在朝向P1 ′-P2 ′ 蛋白结合区的正静电场与抑制活性之间建立了明确的关系。这代表了该系列现在有了一个重要的优化手段,避免了亲和力由P1单元所主导。图5是一些化合物的静电等势面图,显示出了这种特定的效果。
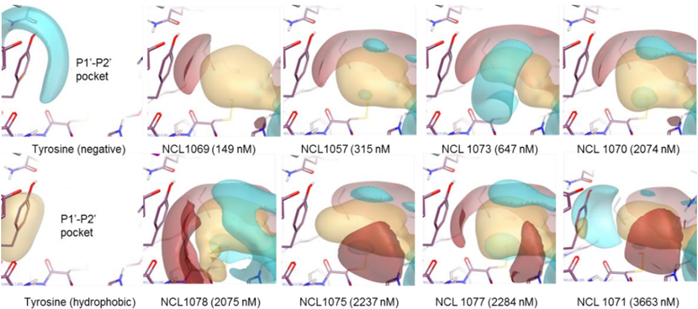
图5. 酪氨酸残基的疏水势(棕色)和负静电势(蓝色)的等势面图(左)。一系列新型matriptase抑制剂的P1′-P2’口袋相互作用基团的正静电势(红色)、疏水势(棕色)和负静电势(蓝色)等势面图(按IC50从左到右递减顺序)。
上图显示了活性如何与配体正等静电势面的厚度(即该特征与酪氨酸残基表面之间的静电互补强度)和立体相互作用的组合有关。酪氨酸芳环的表面有一个相似、显著但符号相反的负静电势表面。在本轮资助结束后,我们得到最有利的化合物是NCL1066,其特性详见表1。
表1. 化合物NCL1066的特性
| Property criteria | NCL 1066 |
|---|---|
| IC50(\( \lt \)0.1μM) | 16nM |
| Ki(\( \lt \)1μM) | 7.8nM |
| Selectvitity (fold over matriptase):(Hepsin,Thrombin,Matriptase-2,Trypsin)\( \gt \)10 | 149x,173x,426x,13x |
| HLM | \( \lt \)1μL/mL/mg protein |
| H plasma stability(\( \gt \)50% rem/2h) | 100% rem/2h |
| Caco A-B flux(Papp)(no significant efflux observed) | 0.32×10-6cm/s (ER ~ 1.33) |
| CYP450 inhibition:(3A4,2C9,2C19,2D6,1A1) (\( \gt \)10μM) | 全部的IC50\( \gt \)25μM |
| Acute cyctotoxicity, PBMC’s | EC50=54.8μM, MEC=39μM |
| Herg IC50 | \( \gt \)5μM |
| MW (\( \lt \)500) | 502 |
| cLogP(\( \lt \)5) | 2.8 |
| HBA(\( \lt \)10) | 9 |
| HBD(\( \lt \)5) | 7 |
| tPSA(\( \lt \)150) | 145 |
| Novelty (patentable) | Novel IP |
结论
从专利突破发展到结构新颖、并具有选择性的系列化合物发现、并最终解决了与靶标相关的许多问题,该项目进展相对较快。其中”膜渗透 “问题从一开始就存在,至今仍有待解决。但是,随着一个强活性系列的建立,通过虚拟筛选具有潜力的第二系列有待开发,该项目已经走上了兑现承诺的轨道。
Cresset的建模为项目提供了独到和极具有价值的见解,并因为它的应用推进了系列1和2的开发而继续为项目增值。
致谢
Sygnature (化学):L. Duffy, P. Meghani;
Newcastle大学 (生物): Prof. Drew Rowan, W. Hui, D. J. Wilkinson, A. Destrument, S. Watson;
Cresset (模拟):A. Vinter.
参考文献
- Ferral, W. R. et al, Protease-activated receptor-2: a novel pathogenic pathway in a murine model of osteoarthritis, J. Arthritis & Rheumatism, 2010 (http://strathprints.strath.ac.uk/20137/).
- Rowan, A. D. et al, Matriptase Is a Novel Initiator of Cartilage Matrix Degradation in Osteoarthritis, Arthritis & Rheumatism, 2010, Vol. 62, No. 7, pp 1955–1966.
- http://merops.sanger.ac.uk/; search: ID = S01.302: Name = matriptase
- Steinmetzer, T. et al, Secondary Amides of Sulfonylated 3-Amidinophenylalanine. New Potent and Selective Inhibitors of Matriptase, J. Med. Chem. 2006, 49, 4116-4126


