
摘要:Pyflare是一个专用的python二进制文件,可以从命令行python脚本中访问Flare的全部功能。这使得诸如对接和EC之类的Flare计算可以在命令行界面上执行,而无需在GUI中加载、存储配体分子和蛋白。随着GUI容量限制的取消,可以自由地在更大的化合物库上执行Flare计算。本文以基于分子对接的虚拟筛选为例,将向您介绍如何编写Pyflare脚本来执行分子对接和EC计算的苗头化合物识别工作流。
作者:Oliver Hills/2023-04-14
编译:肖高铿
背景
在湿法实验室中评估数以百万计的化合物对一个普通蛋白靶标的结合亲和力存在诸多挑战。即使有先进的实验室机器人和自动化平台,湿法实验高通量筛选(HTS)仍然受到时间、资源和资金上的阻碍。
因此,现在普遍的做法是,首先借助于基于结构的药物设计(SBDD)软件平台,试图过滤掉那些被预测为对感兴趣的靶标结合亲和力较低的化合物。这反过来又节省了时间、资源和资金,因为随后的湿法实验室筛选只考虑我们认为最有希望的化合物。
Flare™是一个药物设计软件平台,可用于预测化合物是否会与给定的蛋白靶标结合,使用的技术包括分子对接和静电互补性™(EC)分析,该分析将推算的结合配体与靶标蛋白的结合位点进行的静电特性比较。Flare GUI可以对最多10,000个配体无缝地运行这种计算,但是当我们有数百万个配体时,会发生什么?在这种情况下,不可能在GUI中存储所有这些结构并运行计算。这时,使用Pyflare命令行就会非常有用。
Pyflare是一个专用的python二进制文件,可以从命令行python脚本中访问Flare功能。这是难以置信的强大,因为它使得诸如对接和EC之类的Flare计算可以在您的计算集群的命令行界面上执行,而无需在GUI中加载、存储配体分子和蛋白。随着GUI容量限制的取消,我们可以自由地在更大的化合物库上执行Flare计算。
本文将向您介绍如何编写Pyflare脚本来执行分子对接和EC计算的苗头化合物识别工作流。Pyflare脚本的真正好处是它的可迁移性:该脚本可用于不同的药物发现靶标或化合物库上,而不必进行改变。要启动这个工作流,您只需要三个文件:您的蛋白质靶标存储为PDB文件,共晶的配体存储为SDF文件,需要进行计算HTS测试的化合物也存储在单个SDF文件中。
那么,让我们看看这样一个苗头化合物识别工作流编码成Pyflare脚本是什么样子。
初始化项目
我们需要做的第一件事是创建一个全新、空白的Flare项目。
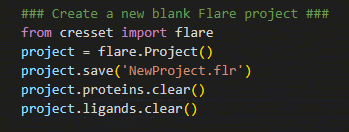
图1.用Pyflare创建一个新的Flare项目
在图1所示的示例中,首先导入Flare软件库,使我们能够访问Flare的计算方法;然后创建一个Flare项目,并将其赋值给变量“project”。通过将Flare项目赋值给变量,使得用户可以执行诸如保存进度(即使用python执行“File | Save As”)、清除当前项目数据(即使用python执行“File | New Project”)以及读入和导出结构文件等操作。
载入蛋白与共晶配体的数据
现在我们已经创建了一个新的Flare项目,让我们首先将准备好的蛋白和共晶配体加载到该项目中。这些结构文件位于我们执行Pyflare脚本的工作目录中。
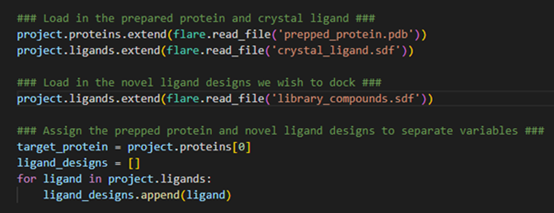
图2. 将蛋白与配体载入到Flare项目中
proteins.extend() “和 “ligands.extend() “操作使我们能够将准备好的蛋白靶标和共晶配体加载到Flare项目中。从概念上讲,我们是通过添加这些新结构来 “扩展”当前的配体和蛋白表单。由于这些新结构存储在不同的文件中,我们需要告诉Flare使用 “flare.read_file() “命令来读取这些文件。对于存储我们化合物库的SDF文件也要做完全相同的流程处理。
现在,苗头化合物识别工作流所需的全部结构都已加载到Flare项目中,我们现在将靶标蛋白和化合物库赋值给不同的变量,以用于后续计算。在这个例子中,只有一个蛋白被加载到Flare项目中,它占据了蛋白表单的第一个数据条。因此,我们可以通过调用蛋白表单的0编号(即表单的第一个条目)索引来给变量 “target_protein”进行赋值。对于Pyflare中的对接和EC计算,用于计算的配体必须以列表的形式存储。因此,通过创建’ligand_designs’变量作为一个空列表,我们可以使用for循环来迭代所有加载到Flare项目中的配体,将每个配体赋值到列表变量’ligand_designs’。现在配体已经以正确的格式存储,以便于计算。
准备配体用于对接计算
苗头化合物识别工作流的下一步是确保我们的配体在进行对接计算之前处于一个能量极小点的3D构象。这可通过进行一个pop-to-3D计算、接着进行一个能量最小化计算而得,如图3所示。
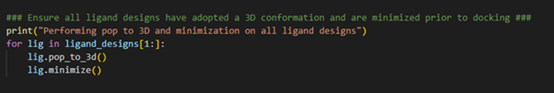
图3. Pop-to-3D与配体结构能量最小化以便用于分子对接
通过建立一个for循环,我们可以循环遍历存储在’ligand_designs’列表变量中的所有配体设计,并执行pop-to-3D和最小化方法,以获得每个配体设计的低能量3D构象。注意 “ligand_designs[1:]”的使用,它允许我们跳过 “ligand_designs “列表变量中的第一个条目。回顾一下,共晶配体是”ligand_designs”列表变量中的第一个条目,因此不需要转换为3D结构。
对接实验
现在,配体设计已经采用了低能量的3D构象,我们可以开始HTS实验的第一个步:对接(图4)。
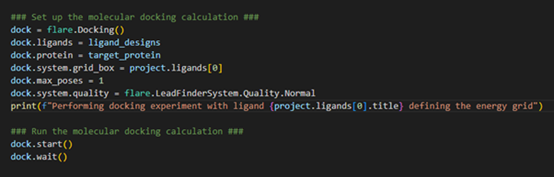
图4.设置对接实验,我们的输入配体将被对接到靶标蛋白上。
我们首先使用’flare.Docking()’加载对接类来初始化对接操作。指定我们希望对接的配体(例如,我们上面定义的’ligand_designs’)与蛋白。在Flare中配置对接计算时,我们还需要定义能量格点的盒子。格点盒子应该可以包住靶标蛋白的活性位点,因此可以被指定为容纳共晶配体的3D空间区域。这是是我们加载到Flare项目中的第一个配体,因此是配体表单中索引号为0的条目(也就是第一条)。在这个例子中,我们仅对保存配体最稳定的结合模式感兴趣,这可通过将“max_poses”设置为1来实现。我们可以通过加载预定义的预设容差(tolerance)来定制对接计算的质量。在本例中,我们选择了“Normal”容差。我们现在可以开始对接计算并等待结果。
计算对接结合模式的静电互补性(EC)
一定对接完成,下一步是计算每个对接结合模式的EC打分。
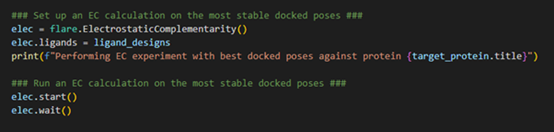
图5. 计算对接结合模式与靶标的静电互补性
对接计算完成后,Flare项目中的每个配体现在被储存为最稳定的对接结合模式,并与蛋白靶标相关联。因此,在初始化EC计算时,通过使用“flare.ElectrostaticComplementarity()”加载静电互补类,我们只需要将Flare指向具有执行EC计算所需的所有相关信息的ligand_designs变量。现在,我们可以启动EC计算并等待结果。
对结果进行分类
工作流的最后一步是确定哪些化合物有望成为蛋白靶标的配体。这是通过对接和EC打分对化合物库进行分类来实现的。明确地说,我们对基于LF Rank Score(在对接实验中计算而得)和EC Score(在EC实验中计算而得)的分类感兴趣。一个具有良好的LF Rank Score和EC Score打分的分子有可能是靶标的强结合配体,而这些就是我们希望从数据集中分离出来的配体。
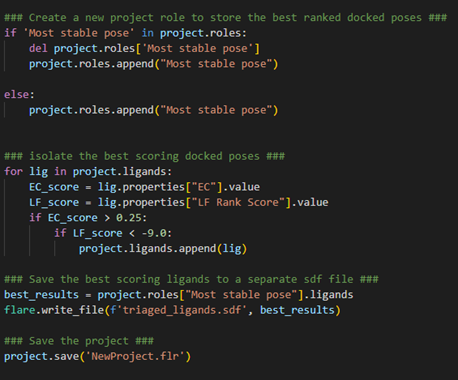
图6. 分析、提取打分最佳的结果
首先,让我们创建一个新的项目角色(project role),恰如其分地命名为“Most stable pose”,以保存提取出来的配体。我们可以先检查这样的角色是否之前已被创建过,如果是,就删除之并重新创建。否则,就用“project.roles.append()”正常创建这个角色。现在,对于我们项目中的所有配体,我们希望识别具有良好的EC Score(大于0.25)和良好的LF Rank Score(小于-9.00)的配体。请注意,我们在这里使用了一个嵌套的if语句,以确保我们捕捉到的配体满足这两个分类标准。对于我们在项目中发现的每个满足这些标准的配体,我们可以将其保存在上面创建的“Most stable pose”角色中。
最后,我们用“lare.write_file()”将分类好的配体导出为SDF文件,并将项目保存以便我们以后再用。
结论
在本文中,我们执行了一个苗头化合物识别工作流,用Pyflare二进制在python中编写脚本,对一个非常大的化合物库进行了计算HTS,并确定了哪些配体有希望成为靶标蛋白的良好结合剂。这都是在不需要在GUI中加载和存储结构的情况下完成的。这个工作流是完全自动化的,计算速度快,而且不受GUI(数据)存储的限制,所有这些都是由Flare的python扩展实现的。
联系我们
联系我们,申请对Flare进行免费试用,以进一步探索其全部的分子模拟能力。


