
摘要:kNN方法是一种著名、健壮的距离学习方法,当传统的2D与3D-QSAR方法建立不出QSAR模型时采用kNN QSAR模型无疑是最好的选择。本教程介绍了kNN QSAR模型的基本原理、使用kNN QSAR模型的场景、kNN QSAR模型的建立流程以及模型质量的评估、kNN模型预测新化合物活性的流程以及预测可靠性的评价等。
一. 前言
在Forge里,叠合过的化合物的静电与形状性质可用k-Nearest Neighbor(kNN)方法开发定量构效关系模型(QSAR model)。
kNN方法是一种著名、健壮的距离学习方法,它预测新化合物活性的过程如下:
- 计算新化合物与全部训练集化合物之间的距离(1-相似性);
- 根据上一步计算的距离,选择与新化合物距离最近的k个训练集化合物;
- 用训练集中与新化合物距离最近的k个的加权平均活性作为新化合物的预测活性值。
当传统的2D与3D-QSAR方法建立不出来QSAR模型时,此时采用kNN QSAR模型无疑最好的选择。标准的2D或3D-QSAR不适合建立模型的情况包括:
- 当训练集化合物涉及多个结构系列时
- 当训练机化合物与靶点具有截然不同不同的结合模式时
- 当化合物的生物活性数据来源不同时
- 当生物活性数据分布不够宽时(活性最佳与最差的差异不到3个数量级时)
预测性kNN模型的开发需要使用合适的距离算法、临近化合物的最佳数k、最佳的权重计算方法。在Forge里,相似性既可以用3D(场与形状)相似性方法,也可以用2D指纹图谱相似性方法。其中kNN 3D-QSAR预测性模型将所有的训练集化合物进行分子叠合再计算3D相似性。
kNN QSAR模型的最佳k值通过留一法(Leave-One-Out,LOO)交叉验证来获取:每个训练集化合物依次轮流剔除,用k个最邻近化合物的平均活性作为预测的活性值。用预测的活性值去计算模型的Q2值。不同的加权方式也同时用于选择距离矩阵以评估方法的性能。
具有最高Q2的k值与加权方式作为最优调教建立kNN模型。
二. kNN模型的建立与应用流程
kNN模型的建立与应用流程如Figure 1所示。
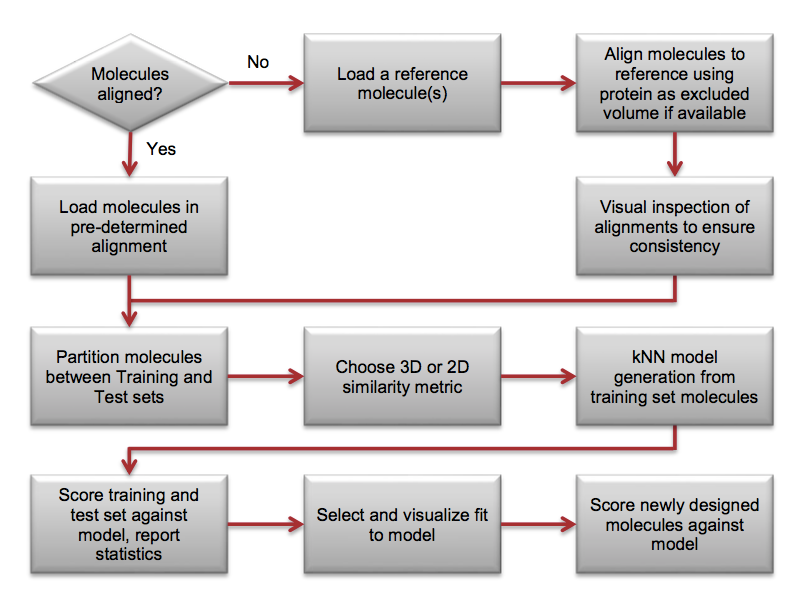
Figure 1. kNN模型的建立与应用流程
三. 建立kNN模型
1. 已经导入训练集与测试集数据,从Process按钮开始
要建立kNN模型,在主工具栏点击’Process’按钮打开处理对话框,然后从Build Model下来菜单里选择’k-Nearest Neighbor(kNN)’,只有训练集分子用来建立模型,但是测试集分子也同时用来预测活性。
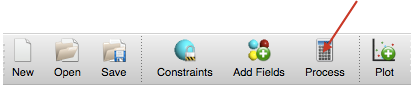
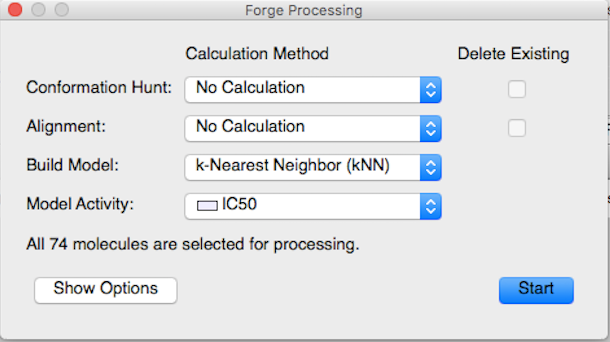
Figure 2. Process按钮
2. 从新建项目开始
还可以以新建项目(File>New Project)的方式从项目导航(New Project Wizard)里选择”Build an Activity Model’,然后再选择’k-Nearest Neighbor (kNN)’, 见Figure 2.
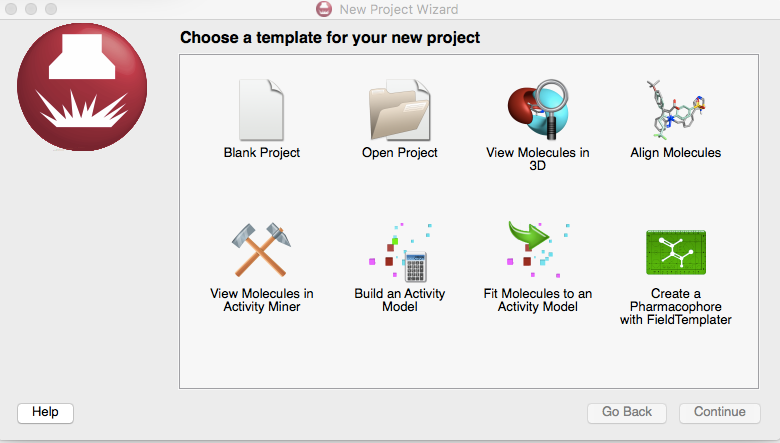

Figure 2. 新建一个kNN活性预测模型
3. 保存模型
保存新建的项目,模型自然保存在项目中,通过读取项目文件来里的模型来预测活性。
三. kNN模型的结果及其质量评估
Forge提供专用的停靠位(dock)界面来展示kNN QSAR模型信息,4个tabs用来展示kNN模型信息,见Figure 3.
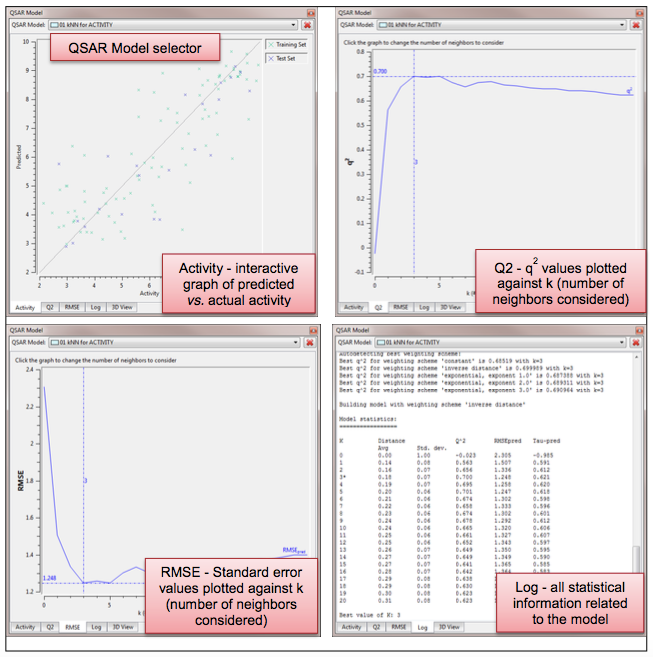
Figure 3. 4个kNN模型的Tabs
1. 活性
活性Tab展示了“预测值-活性值”的相关性图。该图包含了不同系列的数据:训练集,测试集以及预测集。按钮可以展示或隐藏特定的数据集。
活性图中的点可以用鼠标左键拖动画方框来选择,被选中点对应的分子同时也在结果表单中被选中。如果’Show select’按钮处于激活状态,那么被选中的化合物还出现在3D视窗中。用活性图形可以放拜年的检查特定活性范围内化合物的叠合状态。
Figure 4. Show select按钮
如果Forge不能建立出可靠的kNN模型(通过LOO 交叉验证),则训练集化合物建立模型为一个空模型(null model)。训练集中的每个化合物的活性值等于训练集化合物的平均值,因此预测活性值在Activity tab就呈现为一条直线;测试集化合物的预测活性址也都是平均值,因此也呈现为一条直线。
2. Q2
Q2图形tab展示了模型性能(Q2)随最邻近数k的变化。默认情况下,Forge给出第一个最大值Q2的模型。点击希望观察的位置,可以选择不同的k去建立模型。
3. RMSE
RMSE tab图形展示了LOO交叉验证预测均方根差(root mean square error, RMSEpred)随k值的变化。就像Q2 Tab一样,通过鼠标左键点击可以选择不同的k值。
4. Log
Log tab含有建立模型的各种参数设定与信息。鼠标右键可以对窗口中的文本进行选择、复制然后保存到起来、或添加到项目笔记中去。还有一个”Moldel Statistics”表单,其包含了构建Q2与RMSE的原始数据、以及Kendall’s tau系数(Tau-pred)与加权方式。
5. 3D View
kNN模型只是预测性模型,而非解释性(不能指导化合物的设计),因此没有基于场的3D-QSAR模型那样的具有指导结构设计的3D等值图。
四. 预测新化合物的活性
kNN QSAR模型可以用来预测新化合物的活性。
最简单的活性预测方式是使用活性预测向导(见Figure 2的“Fit Molecules to an Active Model”),在预测过程会提示读入已经保存的模型,也就是保存的含有kNN模型的Forge项目文件。
如果kNN模型是用3D静电场/形状(3D Field/Shape)相似性建立,则在预测的过程中会对待预测化合物进行构象搜索与分子叠合。一旦化合物用kNN模型预测(打分)完毕,每个化合物会给出预测的活性值以及预测的可靠度(分子在模型空间的位置)。
预测的活性值在分子表单的Pred列,该列在默认情况是展示(Show)出来,也可以被隐藏(Hide)。你还可以注意到有一列叫”Dist to Model”。该列的值取决于新化合物是否与训练集化合物的相近程度。Excellet,Good或OK表示新化合物与训练集化合物为很近的邻居,因此预测的活性值时可靠的。比这些糟糕的值意味着新化合物并不比邻训练集化合物,因此预测结果完全不靠谱。
分子表单的“Error”列评估用来预测活性的k个邻近化合物的活性值分布(离散)情况。0意味着所有的k个化合物具有完全一样的活性值,因此预测的活性值应该是精确的。该值越大,表明k个邻近分子间的活性值差异大,此时预测的活性值的可靠性也降低。
除了新化合物,训练集与测试集里的化合物也给出预测的活性值、Error与Distance to model。不同的项目的不同预测各自生成不同的Predicted activity、Erro与Distance to model。
五. 接下来可以做什么
- 新化合物活性预测
- 如果收集很多的靶标模型,可以用于靶标预测
六. 申请免费一个月的Forge软件测试
六. 联系我们
试用下载:http://www.cresset-group.com/try-a-free-demo


