
摘要:辉瑞的Trujillo等人在 “双水记(a tale of two waters)”一文描述了对hH-PGDS结合口袋的两个氢键网络水分子进行替换与置换实验,结果表明对主水分子(HOH320)的替换设计导致化合物的活性降低几百倍,而对辅助水分子(HOH305)的置换仅导致活性稍有降低。本文用Flare GIST对结合口袋进行了系统分析,结果发现主水分子HOH320是"Happy"水,而辅助水分子HOH305是"Unhappy"水,这可解释“双水记”观察到的实验现象。Flare GIST水分析对实验设计具有显著的指导作用。
Author: Xiao Gaokeng
First edition: July 25, 2021
Revised: May 21, 2025. Based on user feedback, we’ve enhanced hydration site identification to include both density-based sites and explicit sites. This was achieved by further clustering water density.
前言
并非所有的桥接配体与蛋白相互作用的水分子替换或置换都能提高化合物活性。例如,辉瑞公司的Trujillo等人[1]在文章“双水记(A Tale of Two Waters)”中分享了关于hH-PGDS抑制剂先导化合物的例子,尽管尝试进行了水分子的替换和置换,但并未能改善化合物的结合亲和力。
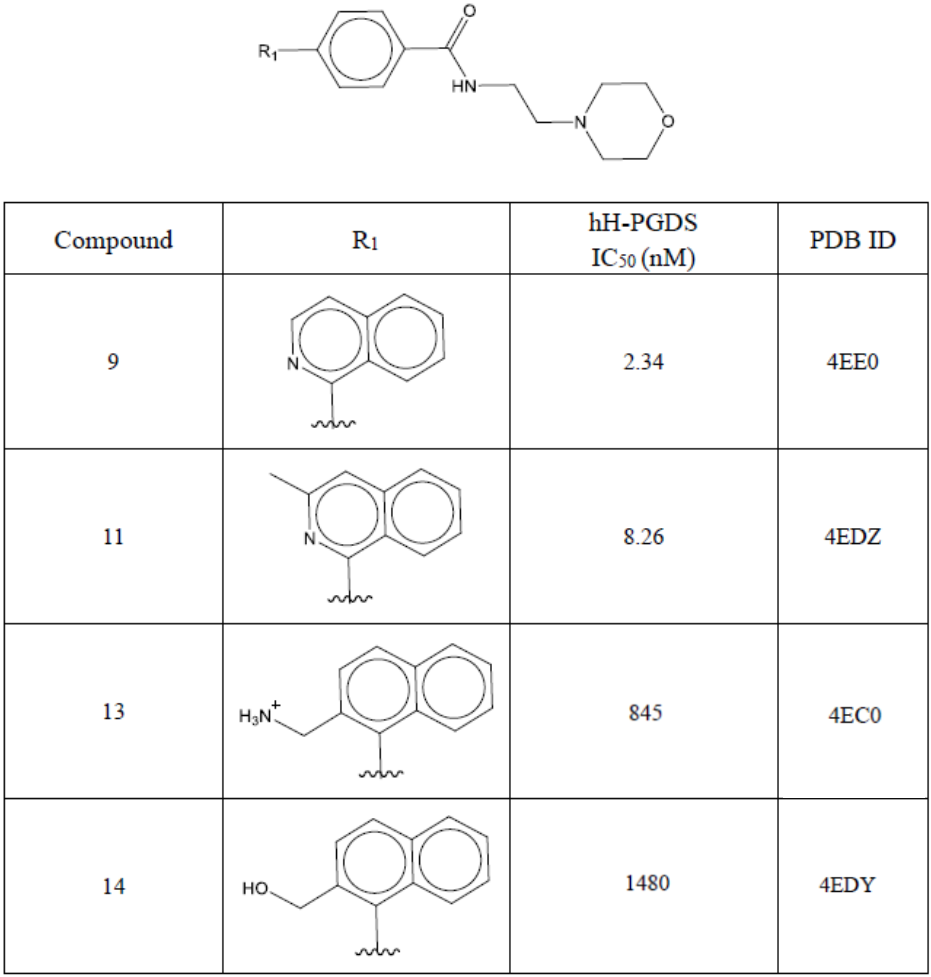
表1. 双水记涉及的4个化合物[1]
观察化合物9(表1)与hH-PGDS的共晶结构(PDB 4EE0)的结合位点,如图1所示,有两个参与氢键网络的水分子HOH320(即双水记之primary water)和HOH305,(即双水记之auxiliary water),它们与化合物9的R1基团有紧密接触。其中HOH320通过氢键网络桥连着配体吡啶环的氮与蛋白LEU199的羧酸氧以及HOH305;HOH305介导了GLY13与其它水分子的氢键网络。Trujillo等人[1]以化合物9为起点分别设计了对HOH305置换的化合物11(表1)、对HOH320替换的化合物13与14(表1)以探讨这种置换或替换对活性的影响。
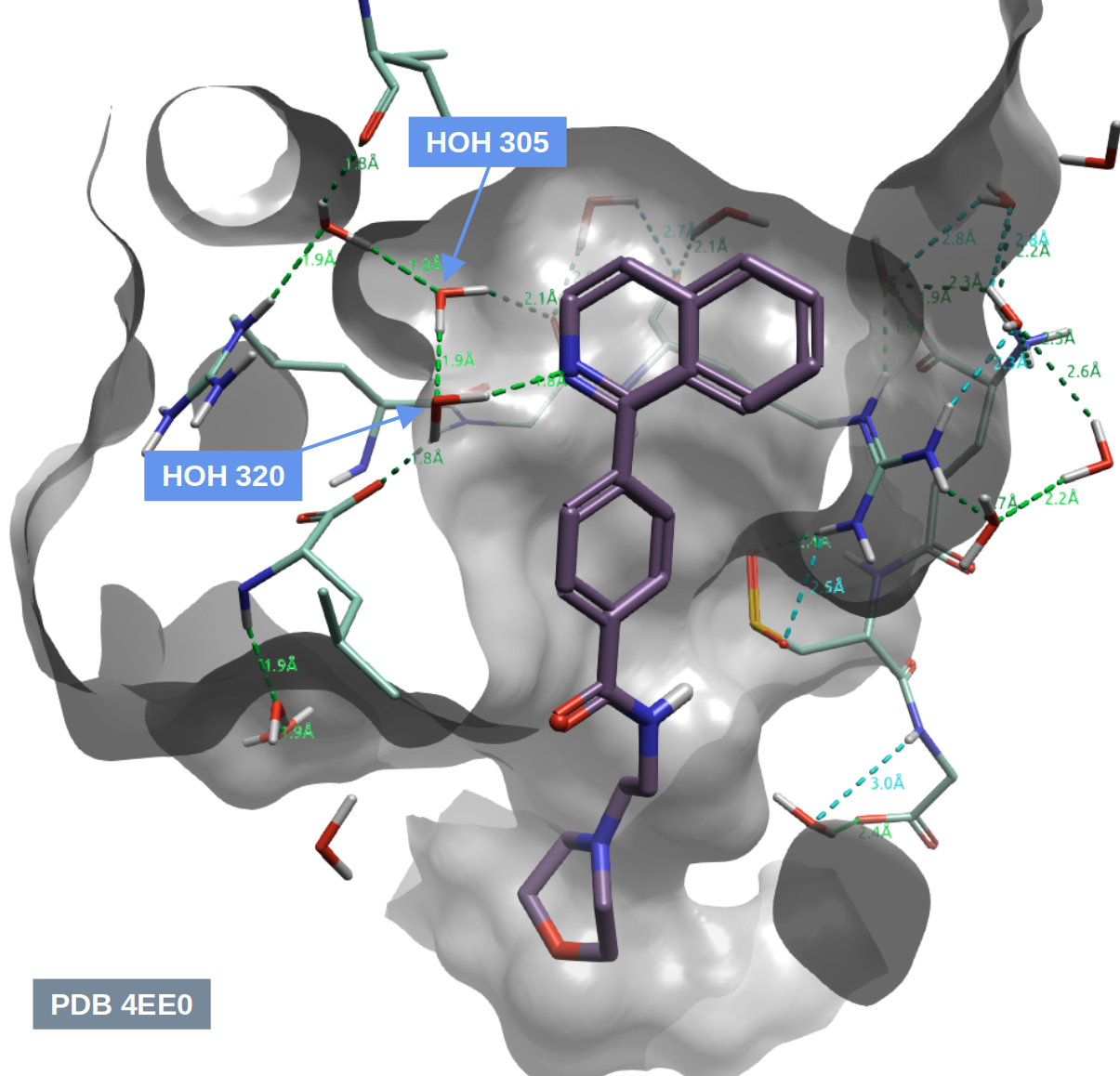
图1. 化合物9与hH-PGDS的共晶结构(PDB 4EE0)的结合模式以及关键的水分子示意图。其中HOH320与HOH305在双水记中分别被称为primary water与auxiliary water。
在“双水记”研究中,作者设计了一个实验:通过在化合物9的吡啶氮邻位引入甲基(得到化合物11),试图置换结合位点中的水分子HOH305。如图2所示,化合物11与hH-PGDS的共晶结构(PDB 4EDZ)显示,该甲基确实占据了HOH305原有的位置。然而,甲基无法像水分子那样参与氢键网络的形成,因此这属于水分子”置换”(displacement)而非功能性”替换”(replacement)。虽然HOH305因此从结合位点消失,但另一个水分子HOH320维持的氢键网络仍然存在。与化合物9(IC50=2.34nM)相比,采用水分子置换策略设计的化合物11对hH-PGDS的活性不仅没有提升,反而下降了3.5倍(IC50=8.26nM)。此外,通过对比PDB 4EE0与4EDZ结构还发现,为适应新引入的甲基,化合物11的分子构象相比化合物9发生了轻微旋转。
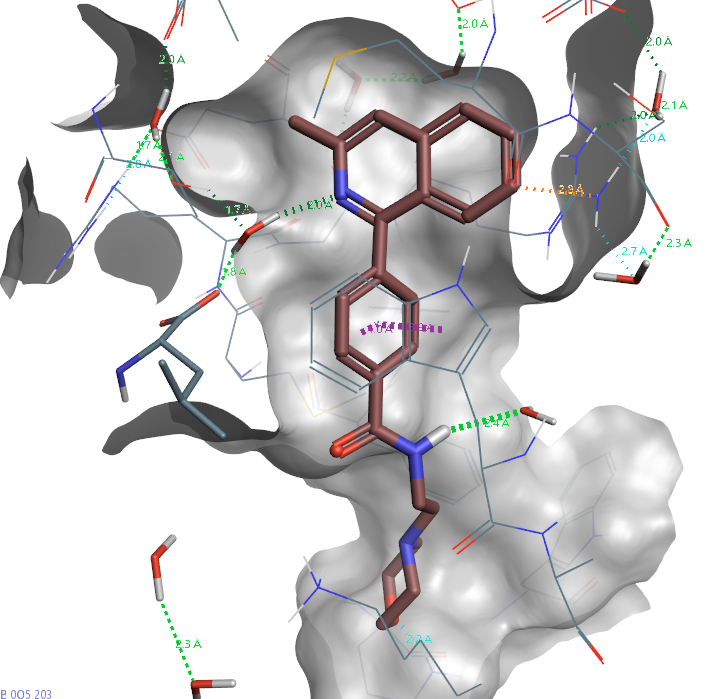
图2. 化合物11与hH-PGDS共晶结构(PDB 4EDZ)的结合模式
在“双水记”的另一项实验中,研究人员尝试替换水分子HOH320。具体策略是将化合物9的吡啶氮原子替换为碳,并分别引入羟甲基和胺甲基,从而得到化合物13和14。这样设计的目的是利用羟基或带正电荷的氮原子来模拟HOH320的作用,并将其整合到新化合物中。如图3所示,化合物13和14与hH-PGDS的共晶结构表明,它们的羟基和胺基确实占据了HOH320的位置,并且成功介导了与HOH320相同的氢键网络。同时,HOH305原有的氢键网络仍然保持完整。这一结果表明,该设计完美实现了水分子替换策略。
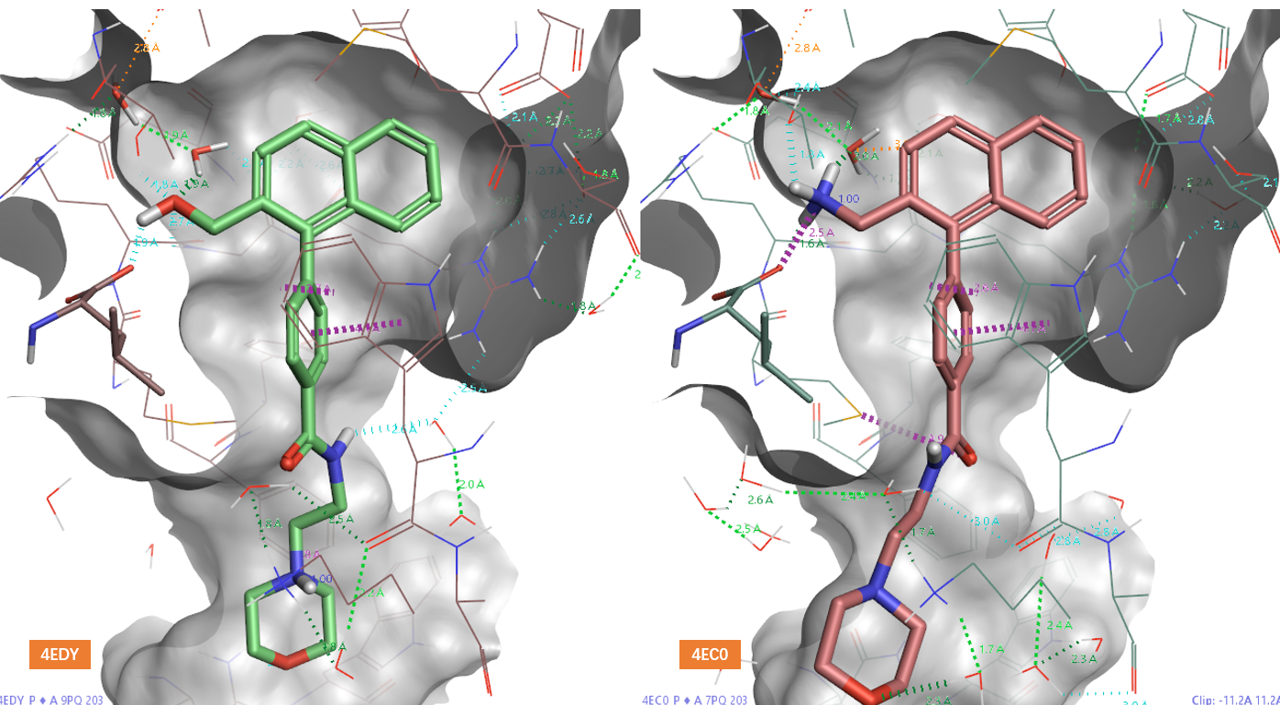
图3. 水分子替换策略设计的化合物13、14与hH-PGDS共晶结合模式。
然而,体外酶学实验结果却出人意料。与化合物9(IC50=2.34nM)相比,采用HOH320替换策略设计的化合物13和14对hH-PGDS的抑制活性不仅没有提升,反而分别大幅下降了360倍(IC50=845nM)与630倍(IC50=1480nM)。这一结果与HOH305置换策略设计的化合物11(活性仅下降3.5倍)形成了鲜明对比。
晶体结构分析表明,化合物13和14中连接杂原子(O/N)与萘环的连接臂二面角接近平面构型(分别为21°和27°),而优势二面角应为90°。此外,其萘环与苯环连接臂的二面角也偏离低能构象约20°(以化合物14为例,实测117° vs 低能构象97°)。作者认为,这种显著的构象张力导致分子内能升高,其能量代价可能远超溶剂置换带来的熵增收益,这很好地解释了化合物13和14活性显著降低的原因。
Trujillo等人[1]的一个重要结论是:要获得对hH-PGDS的最佳结合亲和力,抑制剂必须与活性位点中的关键水分子HOH320形成氢键相互作用。结构生物学数据表明,HOH320所处的微环境具有严格的几何和化学限制,使得用抑制剂原子直接置换该水分子的策略难以实现。相比之下,复合物对次要水分子HOH305的置换表现出良好的耐受性,仅导致抑制剂结合亲和力轻微下降(约3.5倍)。
除了结合构象的张力能会影响化合物的活性之外,结合位点水的热力学性质是另一个影响活性的因素。由Nguyen等人[2]提出的GIST(Grid Inhomogeneous Solvent Theory)方法使用显式水对蛋白质进行约束的分子动力学模拟,并使用非均匀溶剂化理论 (inhomogeneous solvation theory,IST) 计算预定义体积(通常是结合位点)的水分子分布和热力学性质。本文的主要目的是用Flare™ [3]实现的GIST系统地分析HOH320和HOH305的水分子热力学性质,从“水”的角度考察Trujillo等人[1]水分子置/替换策略没有达到预期提供理论解释。
结果
apo-GIST分析
图4. 化合物9与hH-PGDS共晶结构(PDB 4EE0)结合位点的apo-GIST水分析结果。其中白色网格等值图:Water Density \(\geq\) 4;红、绿色球状分子:apo-GIST预测的水合位点;分子表面:PDB 4EE0的hH-PGDS;细棍状:共晶水与配体。
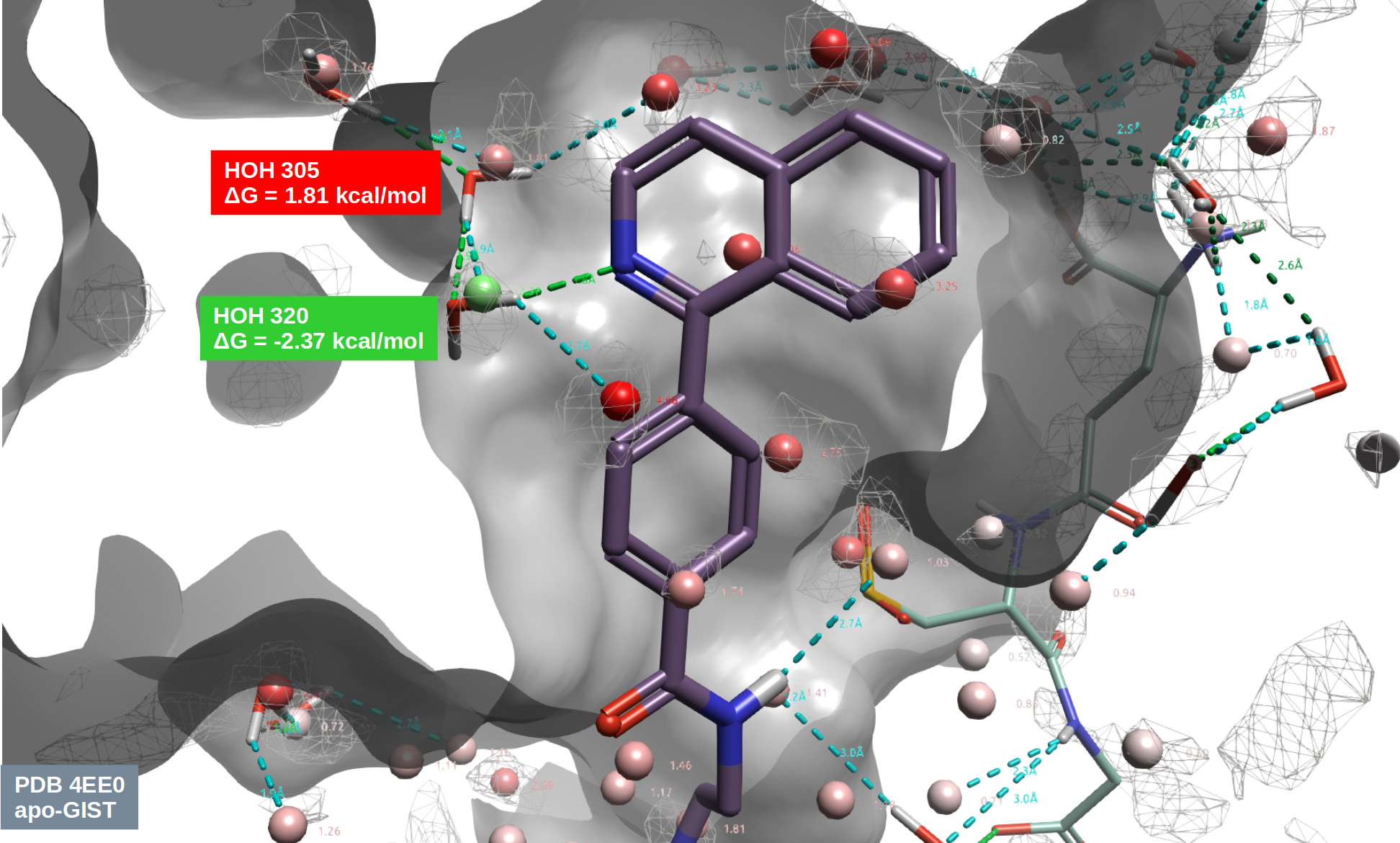
用Flare GIST对化合物9与hH-PGDS共晶结构PDB 4EE0结合位点进行的apo-GIST分析结果如图4所示:其中白色网格为Water Density \(\geq\) 4的等值图;红、绿色球状分子为apo-GIST预测的水合位点;深灰色分子表面表示hH-PGDS;细棍状为共晶水与配体。GIST的水密度即是预测的水合位点,可以看到,包括HOH320与HOH305在内的共晶水被Water Density \(\geq\) 4的水密度图覆盖,这说明GIST预测水合位点的精度相当高。
此外,还可以对水密度进行聚类分析得到显式的水合位点。首先,选择具有最高水氧密度的体素来确定第一个水合位点的位置。然后排除所有在第一个水合位点2.5Å范围内的体素,不再考虑。然后,对下一个最高密度的体素重复此过程,直到没有剩下密度高于2倍本体水氧密度的体素为止。图4的球状分子就是根据水密度计算得到的水合位点,可以看到,水合位点都是位于密度图里面。注意到,HOH320与HOH305分别与两个绿色、红色的水合位点重合,这进一步证明了GIST水合位点预测的准确性。其中水合位点的颜色是根据GIST计算的水合自由能进行着色。
由于GIST算法[2]将IST中的熵和能量表达式出现的空间积分离散化到一个细致、连续的三维网格(体素)上,因此为了计算水合位点的自由能需要将水合位点占据的体素的自由能进行加和。图4的ΔG即是加和后的结果。可以发现,与HOH320重合的绿色水合位点ΔG = -2.37 kcal/mol,是个“Happy”水;而HOH305处红色水合位点ΔG = 1.81 kcal/mol,是个“Unhappy”水。这解释了表1的SAR,化合物13、14因为替换Happy的HOH320而比化合物9活性大幅降低;而置换HOH305的化合物11从能量上看是有利的,但是这个置换没有“继承”到HOH305的氢键网络,因此活性没有增加,而是温和地下降了。
holo-GIST分析
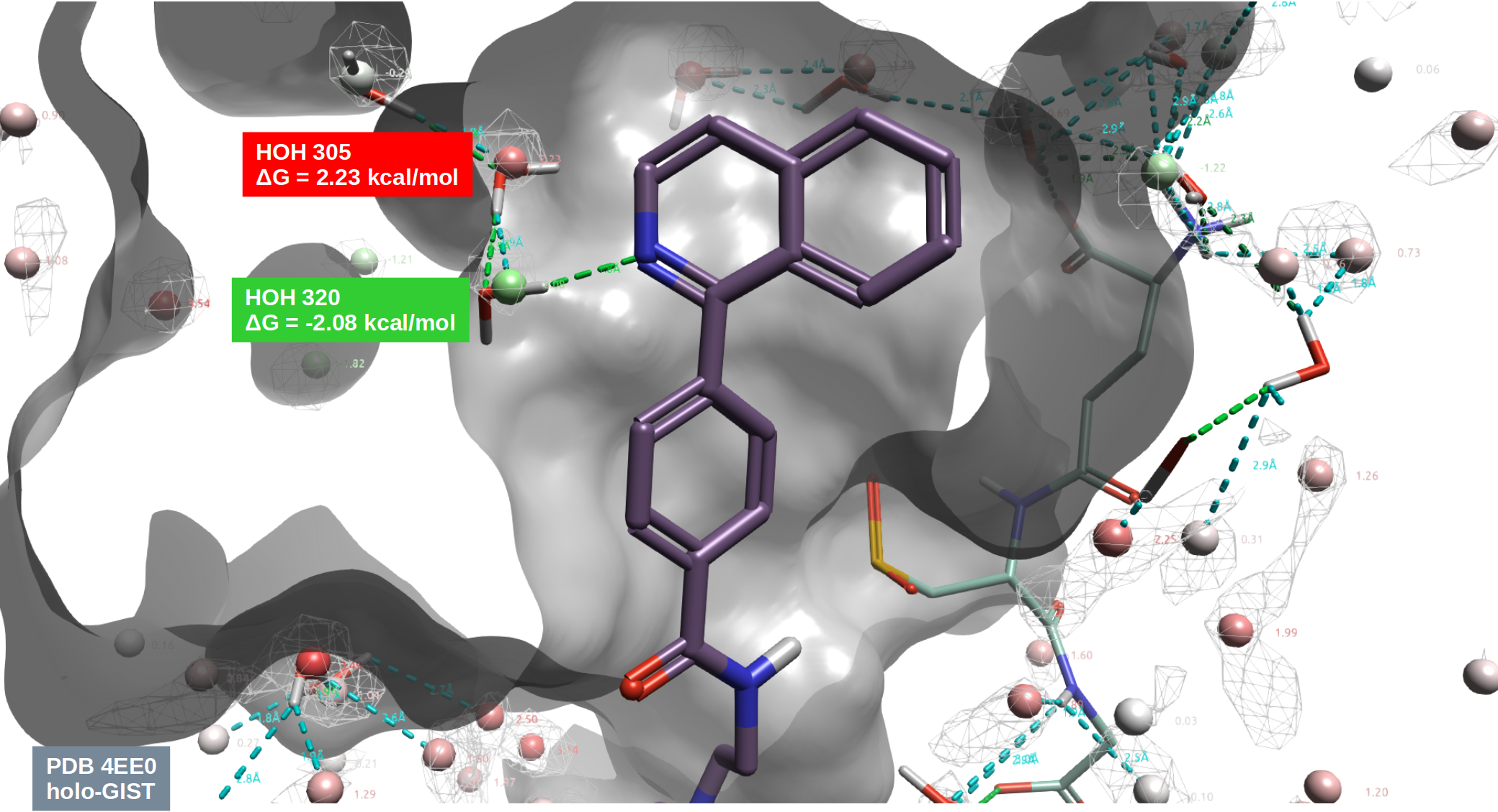
图5. 化合物9与hH-PGDS共晶结构(PDB 4EE0)结合位点的holo-GIST水分析结果。其中白色网格等值图:Water Density \(\geq\) 4;红、绿色球状分子:holo-GIST预测的水合位点;分子表面:PDB 4EE0的hH-PGDS;细棍状:共晶水与配体。
为了进一步确认HOH320在介导化合物9与hH-PGDS相互作用的重要性,还对结合口袋进行了holo-GIST分析,结果如图5所示: HOH320在复合物结合位点里是happy水,ΔG = -2.08 kcal/mol。这说明了HOH320的存在稳定了化合物9与蛋白之间的相互作用而对结合亲合力有利,这与Trujillo等人[1]的结论一致,即抑制剂必须与HOH320形成氢键相互作用才能获得对hH-PGDS的最佳结合亲和力。在化合物9与hH-PGDS的holo结合口袋里,HOH305是Unhappy水,ΔG = 2.23 kcal/mol,因此对这个水的置换设计是能量有利的。
总的来说,用Flare GIST对化合物9结合位点的分析表明,HOH320是happy水,应当在设计中予以保留或者用配体的一部分进行替换以模拟水的作用;HOH305是unhappy水,可以被置换或替换。这与观察到实验结果基本一致,对happy的HOH320替换设计化合物13与14活性大幅降低了几百倍,而对unhappy的HOH305进行置换设计的化合物11仅在活性上稍微降低(降低了3.5倍);这也与Trujillo等人[1]的结论一致。在本算例中,Flare GIST水分析计算对实验设计具有正确的指导作用。
方法
结构准备
根据PDB代码将晶体结构从蛋白质数据库下载到Flare V103中,并使用来自Protein Prep工具小心地准备,以添加氢原子、优化氢键、消除原子冲突并将最佳质子化状态分配给蛋白质结构。任何截短的蛋白质链被封端作为蛋白质准备的一部分。
在Flare中Sequence/align多重比对工具比对蛋白质序列,然后通过Cα的最小二乘拟合进行叠合,结合口袋的比较都是基于这个叠合来实现。
GIST计算
以化合物9共晶结构PDB 4EE0的apo-GIST计算为例,将蛋白下载到Flare V103,然后进行结构准备,最后共晶结构的A链水包含在GIST分析里面,具体的GIST分析条件如下:
- Calculation method: Normal
- Ligand: None
- Grid spacing: 0.5 Å
- Grid Definition:Ligand
- Chains: A Chain, A Ligand,A Other,A Water
- Simulation length: 20ns
- Solvent Model: explicit TIP4Pew Water
- GCNCMC: During equlibration only with buffer 4.00 Å
在这个计算中,在分子动力学模拟之前将共晶结构的水链包含在GIST分析里,并将配体GSF与金属Mg考虑在内。计算完毕,将水密度(Water density)以及ΔG导出为dx格式文件用于一下步的水合位点预测与水合自由能计算。
holo-GIST采样了同参数,所不同的是Ligand不为None,而是共晶配体A 0O4 202。
水合位点预测与水合自由能计算
对水密度进行聚类分析得到显式的水合位点。首先,选择具有最高水氧密度的体素来确定第一个水合位点的位置。然后排除所有在第一个水合位点2.5Å范围内的体素,不再考虑。然后,对下一个最高密度的体素重复此过程,直到没有剩下密度高于2倍本体水氧密度的体素为止。计算采用基于pyflare的脚本gist_hydrate_sites.py来实现4。
然后识别每个水合位点周围1.4Å半径内的体素(Voxel):
$$
V = \left\{v_{i} | v_{i} \in hydration\ site\right\} \cdots(1)
$$
如方程1所示,如果一个体素包含在水合位点的范德华半径内,则累加这些体素的能量,并将总和乘以体素的体积(vol = 0.125 Å3),得到以kcal/mol为单位的值,如方程2所示:
$$
ΔG = vol \times \sum_{v_{i} \in V}G_{GIST}(v_{i})\cdots(2)
$$
方程2的ΔG即是水合位点的水合自由能。在计算的时候,对体素vi上的GGIST值设置了下限截断值±0.5kcal・mol-1・Å-3来去掉贡献不显著的体素,使用上限截断值+3kcal・mol-1・Å-3去掉极端高值的体素,如方程3所示:
$$
G_{GIST}(v_{i}) = \left \{
\begin{aligned}
& 0\ &if\ |G_{GIST}(v_{i})| \lt 0.5 \\
& +3\ &if\ G_{GIST}(v_{i}) \ge +3 \\
& G_{GIST}(v_{i}) &\ otherwise
\end{aligned}
\right.\cdots(3)
$$
结论
辉瑞的Trujillo等人在 “双水记(a tale of two waters)”一文描述了对hH-PGDS结合口袋的两个氢键网络水分子进行替换与置换实验,结果表明对主水分子(HOH320)的替换设计导致化合物的活性降低几百倍,而对辅助水分子(HOH305)的置换仅导致活性稍有降低。本文用Flare GIST对结合口袋进行了系统分析,结果发现主水分子HOH320是”Happy”水,而辅助水分子HOH305是”Unhappy”水,这可解释“双水记”观察到的实验现象。Flare GIST水分析对实验设计具有显著的指导作用。
算例下载
对PDB 4EE0进行GIST计算的Flare算例:PGDS-4EE0-GIST.flr
在你自己的项目中试用GIST
如果您希望在您的项目中试用GIST功能,请立即申请免费评估版的Flare。
- 电邮:info@molcalx.com
- 电话:020-38261356
文献
- Trujillo, J. I.; Kiefer, J. R.; Huang, W.; Day, J. E.; Moon, J.; Jerome, G. M.; Bono, C. P.; Kornmeier, C. M.; Williams, M. L.; Kuhn, C.; et al. Investigation of the Binding Pocket of Human Hematopoietic Prostaglandin (PG) D2 Synthase (HH-PGDS): A Tale of Two Waters. Bioorg. Med. Chem. Lett. 2012, 22 (11), 3795–3799. https://doi.org/10.1016/j.bmcl.2012.04.004.
- Nguyen, C.; Gilson, M. K.; Young, T. Structure and Thermodynamics of Molecular Hydration via Grid Inhomogeneous Solvation Theory. 2011. arXiv:1108.4876v1. https://arxiv.org/abs/1108.4876
- Flare™, version 10, Cresset®, Litlington, Cambridgeshire, UK; https://www.cresset-group.com/flare/; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation J. Chem. Inf. Model. 2006, 46 (2), 665-676; Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes J. Med. Chem. 2019, 62, 6, 3036-3050; Maximilian Kuhn, Stuart Firth-Clark, Paolo Tosco, Antonia S. J. S. Mey, Mark Mackey and Julien Michel Assessment of Binding Affinity via Alchemical Free-Energy Calculations J. Chem. Inf. Model. 2020, 60, 6, 3120–3130
- 基于GIST的水合位点分析及其在基于结构设计中的应用. 墨灵格的博客. Available at: http://blog.molcalx.com.cn/2024/10/04/gist-based-hydration-site-analysis.html


