
摘要:本文以Imatinib、dasatinib与ABL1激酶复合物结合模式为例,解释为什么分子对接并不是一件容易的事。
问题
药化的朋友通常会让我帮忙:“我有一个活性非常好的化合物,请帮我做一个分子对接(预测结合模式),然后帮我做个漂亮的相互作用分析图片”。在一般情况下,这个问题的最重点是“图片”。而我通常会说:“把你的化合物结构给我看看,我要查些文献以确定能不能做”。这时药化朋友通常会很惊讶:“我已经查过文献,PDB里有蛋白结构结构可以下载,你跑个对接计算就可以了”。然而我想说结合模式预测并非只是使用软件跑一遍流程那么简单的事。
正确的蛋白结构是结合模式预测的关键
我们知道,蛋白与配体结合过程会涉及诱导契合,apo结构(不结合配体的蛋白)与holo结构(结合了配体的蛋白)的结合口袋形状可能不同,结合了不同配体的holo结构的结合口袋形状也可能完全不同。单这一点就可以让分子对接结果不靠谱,以下举例说明。
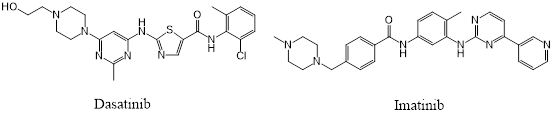
图1. BCR-ABL1激酶抑制剂Dasatinib与Imatinib的化学结构
Dasatinib(图1-左)与Imatinib(图1-右)都是靶向BCR-ABL1激酶的慢性骨髓性白血病治疗药,它们的蛋白-配体复合物结构分别为PDB 2GQG与1IEP。假设我们只有dasatinib-ABL1激酶的复合物结构2GQB(图2-右),我们能否将Imatinib对接到2GQB的Dasatinib结合位点而预测出Imatinib-ABL1复合物1IEP结合模式(图2-左)呢? 这个就是所谓的cross-docking问题[1],已经有很多文献与综述讨论,这里仅是用一个简单的例子来说明cross-docking才是我们真实的对接计算场景。
我们知道激酶的DFG片段通常有两个典型构象-in与out:采取out构象时别构位点处口袋暴露出来,in-构象时别构位点处被堵上。Imatinib与DFG-out构象的ABL1结合,其结合位点口袋形状如下图2-左所示;而Dasatinib与DFG-in构象的ABL结合,其结合位点口袋形状如图2-右所示。
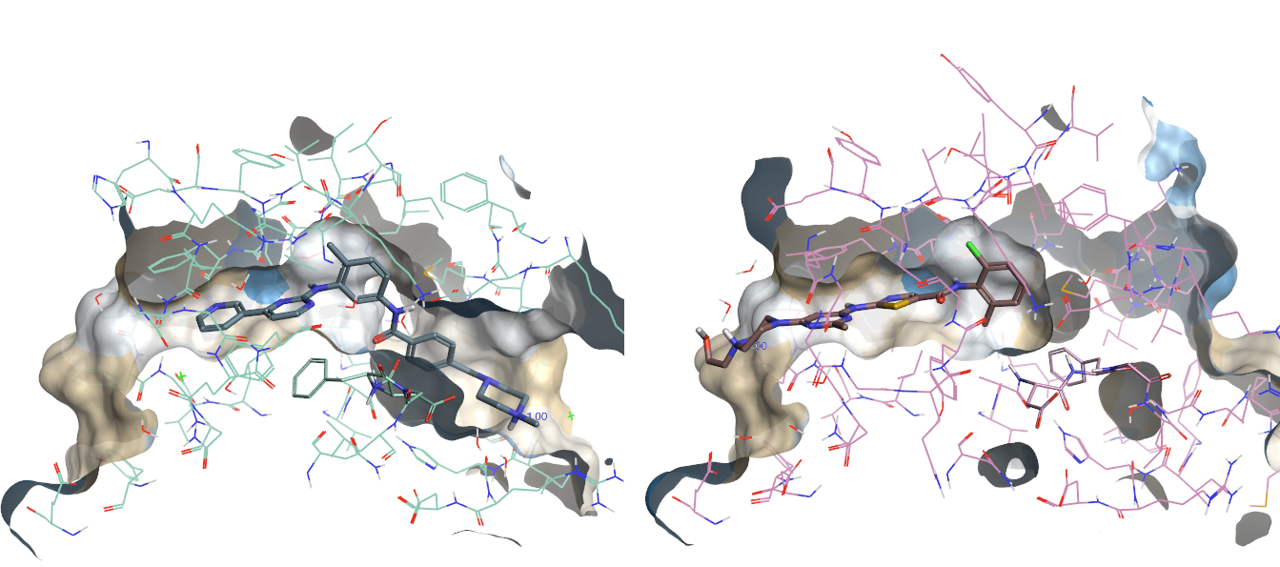
图2. Imatinib(左)与Dasatinib(右)与BCR-ABL1激酶的共晶结构结合位点示意图,PDB代码分别为1IEP与2GQG
如图2所示,右边dasatinib-abl共晶结构的结合口袋在化合物右侧苯环处就堵死了,而左边imatinib相应位置却打开一个疏水通道并与配体匹配。很明显,与dasatinib结合的蛋白口袋由于形状与尺寸的差异而容纳不了imatinib,由此可以预见:尝试将imatinib docking到PDB 2GQG的结合位点里是获得不了正确的结合模式的。那我们平常不加思索地用分子对接将一个化合物对接到随机选择的蛋白结合位点里来预测结合模式,会不会刚好就属于这种用了不适的口袋的情况呢?
系综对接:信息利用的越多,正确的概率越大
为了提高分子对接预测结合模式的成功率,人们想了多种办法:比如柔性蛋白侧链的分子对接;与分子动力学模拟结合使用;充分利用现有信息的系综对接(ensemble docking)等等。这里,我重点聊聊系综对接的策略:将PDB里同一蛋白的所有晶体结构都用来进行对接计算,充分利用所有可获得的蛋白结构信息,以提高正确预测的概率。如图3所示,假设同一蛋白的同一结合口袋已知的复合物结构有”十字型”与”三角型”两种(图3左),您要预测的新化合物结合到同一结合位点的“直角型”结合口袋(图3右)里。
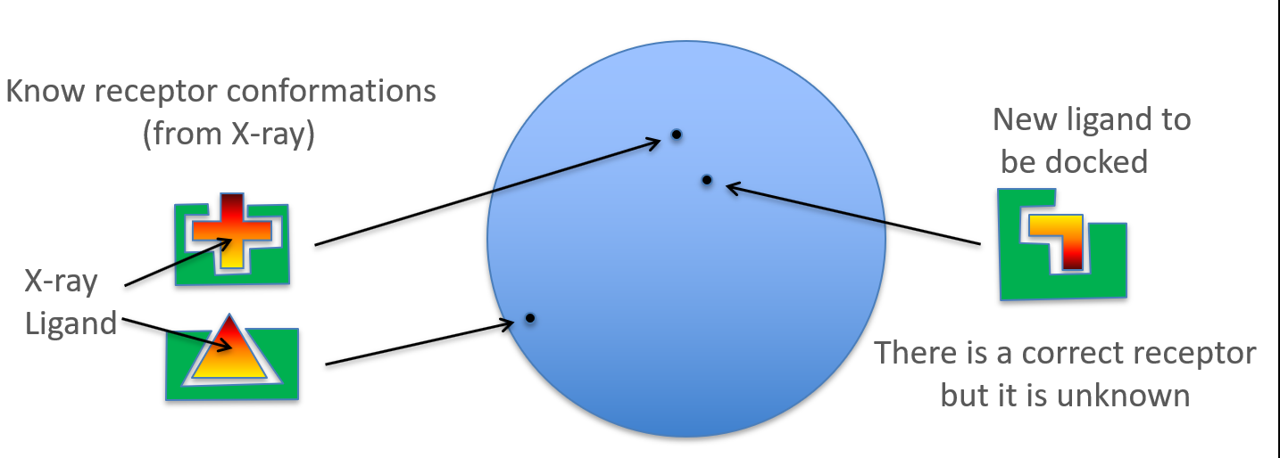
图3. 同一蛋白的同一结合位点与不同配体产生不同形状的结合口袋示意图。
首先,我们要下载该蛋白的所有已知结构,并将蛋白准备成分子对接直接可用的形式,这是个费时费力的过程。有的蛋白有几百个复合物结构,此时工作量非常巨大。如图4所示,我们需要用分子对接软件将直角型的新化合物在所有的蛋白结合位点里进行docking计算、预测结合模式,然后排序、挑选合适的结合模式。
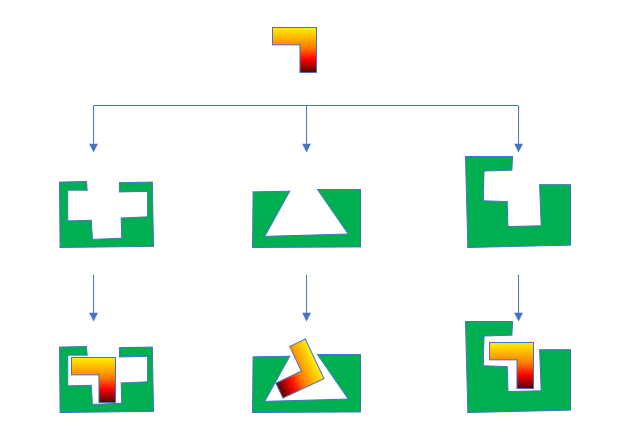
图4. 系综对接(Ensemble docking)示意图
如图4所示,如果与新化合物匹配的“直角形”蛋白结合口袋(图4,右)已经存在于现有的数据库里,那么就有机会预测得到正确的结合模式;如果与新化合物匹配的结合口袋不存在于现有的数据库里,或许有机会命中次选的“十字型”蛋白结合口袋里(图4 左);如果运气不好,只有一个三角形的结合口袋(图4 中)是已知的,那么docking计算预测的结合模式将都是不对的。更值得警惕的是,此时或许对接打分还看着不错(理想情况下对接打分不应该好),则会严重误导对结合模式的选择。总的来说,系综对接策略可以充分利用现有的蛋白信息,让我们有更多的机会在正确的蛋白结合位点里预测到正确的结合模式。
当然,系综对接的蛋白结构除了来源自实验(比如从PDB下载)之外,还可以来源自分子动力学模拟。将分子动力模拟获得的蛋白构象系综用于分子对接或基于结构的药效团虚拟筛选,请参阅另外两篇博客文章[3,4]。
结合模式预测的准确度与结构准备有关系
除了上述正确结合口袋的选择很重要之外,正确的蛋白与配体结构准备也会影响对接结合模式。我们已经很详细地讨论过如何进行蛋白与配体的结构准备,这里就不再详述。
结合模式预测与分子对接打分值
根据侯廷军课题组的一项测试,打分最佳的结合模式为正确的结合模式的概率为40-60%。但是,在打分最佳的20个结合模式里,含有正确结合模式的概率会提高到60-80%。因此,单纯依靠软件决定结合模式的正确率目前还不够高,需要依靠可视化分析(视觉检查、人工检查)来挑选出正确的结合模式[2]。
实验结合亲和力与分子对接打分值
来自实验学科朋友的另一个主要兴趣点是解释两个相似分子的活性差异。关于这个问题,请参见:关于用分子对接预测结合亲和力的问题。
分子对接与虚拟筛选
分子对接虽然不能精确区分化合物的活性高低,但是有能力区分活性的有无(活性差异3个数量级或以上),因此分子对接可以用来从数据库里富集与特定口袋结合的潜在化合物,也就是虚拟筛选。虚拟筛选或许会错过不与当前结合口袋匹配的活性化合物,但是可以高效为项目推荐出苗头化合物或产生想法,加速项目的进度。
文献
- Sutherland, J. J.; Nandigam, R. K.; Erickson, J. A.; Vieth, M. Lessons in Molecular Recognition. 2. Assessing and Improving Cross-Docking Accuracy. J. Chem. Inf. Model. 2007, 47 (6), 2293–2302. https://doi.org/10.1021/ci700253h.
- 基于结构药物发现中的决策:对接结果的手工检查. 墨灵格的博客. http://blog.molcalx.com.cn/2021/05/22/visual-inspection-of-docking-results.html
- 用分子动力学模拟生成蛋白构象系综用于对接实验更具生物学合理性. 墨灵格的博客. http://blog.molcalx.com.cn/2022/10/26/ensemble-docking-md-flare.html
- 药效团模型的分层图表示. 墨灵格的博客. http://blog.molcalx.com.cn/2021/06/06/hierarchical-graph-representation-of-pharmacophore-models.html


