
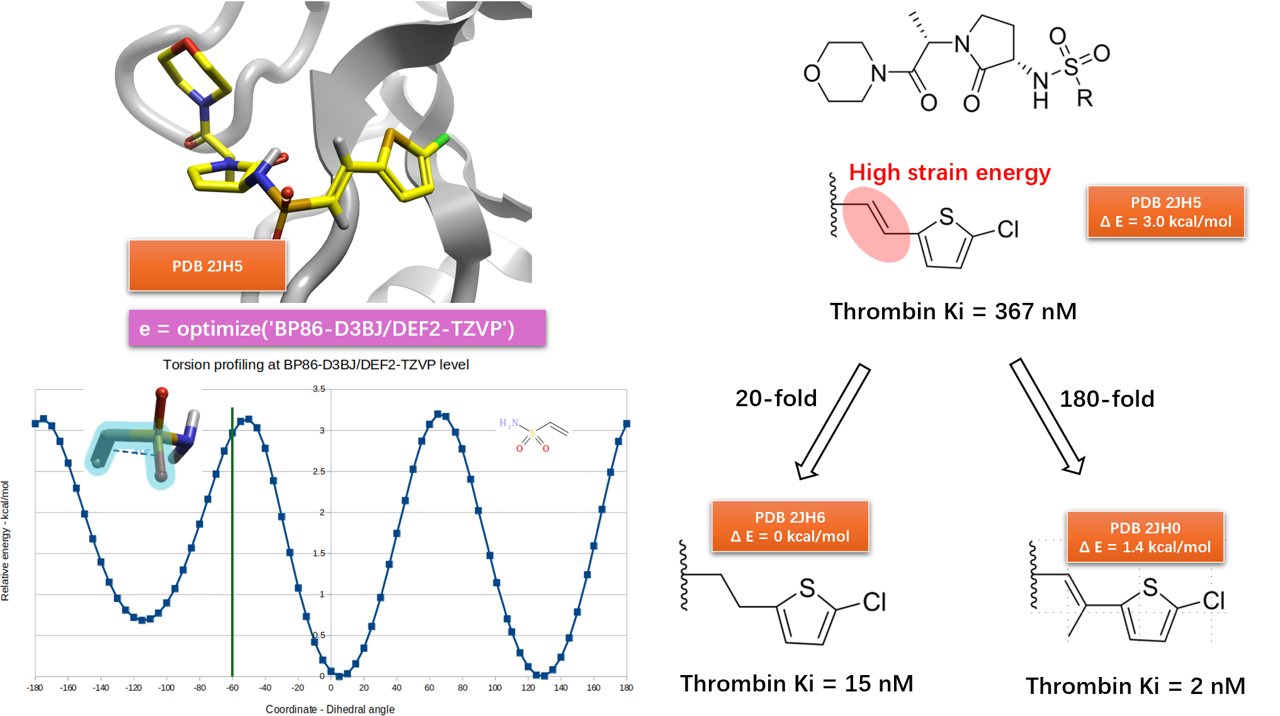
在基于结构的设计中用QM扭转角分析研究构象效应
摘要:GSK的Senger等人结合结构生物学与量子化学计算方法,确认了一对仅一个价键差异的分子,其20多倍的生物活性差异是由构象效应引起的。本文用Flare QM回溯性地重现了文献报道的发现,表明构象效应在分子识别中起着重要作用。在基于结构的设计中,使用QM扭转角分析可以方便、可靠地考虑这类构象效应,并用于指导新分子的设计。
作者:肖高铿/2024-03-03
前言
想要基于结构的设计(structure-based design,SBD)项目取得取得成功,需要将大量的结构生物学信息转化为对分子识别原理的正确理解。由于配体与生物靶标之间的相互作用是一个高度复杂的过程,因此很难以用定量的方式描述其相互作用能。为了简化结合能的计算,通常假设结合能由可以单独计算的不同贡献项累加而来。例如,Ajay和Murcko等人1将结合能分为以下几个部分:(1)形成复合物的反应物间的相互作用;(2)溶剂化/去溶剂化;(3)形成复合物时与生物靶标以及配体的“运动”变化相关的能量变化(即旋转和平动自由度的损失);(4)配体和生物靶标在形成复合过程中的构象变化。
然而基于结构的设计(structure-based design,SBD)是一项具有挑战性的工作,因为即使是局部的SAR也很难通过一个单一主导因素的变化来解释。自从1995年Ajay与Murcko等人1提出计算方法以来,几乎没有结构生物学证据支持构象效应在SBD中的重要性,因为单一构象效应的SAR太少了。直到2007年GSK的Senger等人2在研究选择性FXa抑制剂时为此特意设计了一对分子5与6(图1),并解释了它们与凝血酶(Thrombin)的共晶结构,将结构生物学信息与基于量子化学计算方法的扭转角分析(QM Torsion profile)相结合,确认观察到的大约20倍生物活性差异主要是由构象效应引起。接着作者利用构象效应指导设计了化合物7(图1),扭转角分析预测7比之5降低了构象能,随后的生物活性测试表明7比之5大约实现了180倍地活性提升,这不仅证明了构象效应在SBD中的重要性,并证明其可以作为定性指导在SBD工作中普遍应用。
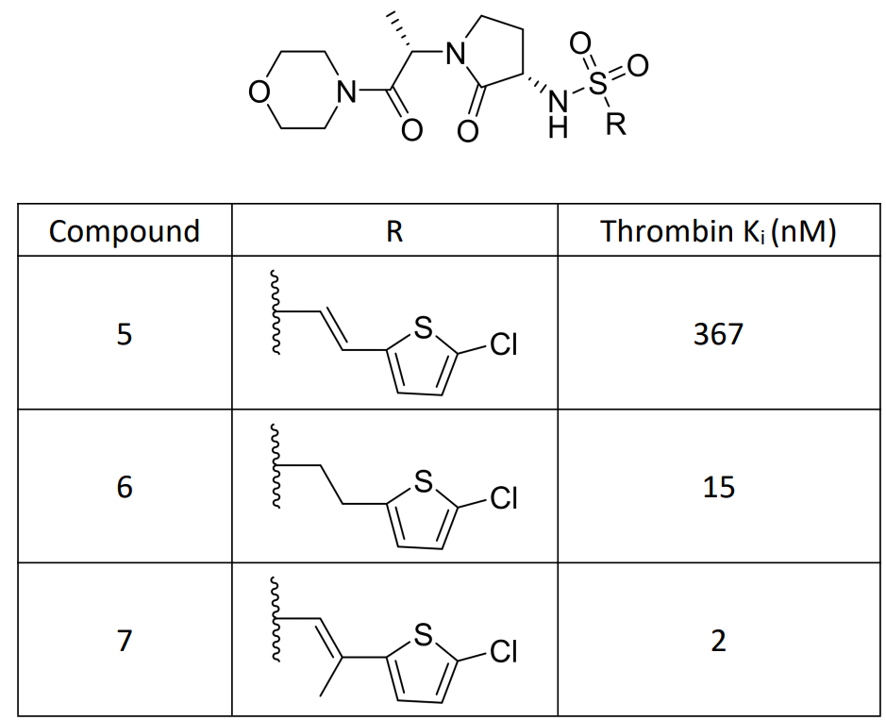
图1. 化合物5、6与7及其Thrombin抑制活性
本文的目的是回溯性地用Flare QM3,4的Torsion Profile来重现Senger等人2的构象效应分析过程,以呈现Flare QM在基于结构设计中研究构象效应的高效性以及使用上的便利性。
材料与方法
蛋白与配体结构的准备
将共晶结构PDB 2JH5、2JH6与2JH0下载到Flare™ V83中,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,并对截短的蛋白质链继进行封端。从准备好的复合物结构里将配体提取出来,得到5、6、7的生物活性构象。
模型分子的准备
模型分子是从上一步准备好的活性构象配体基础上截取得到,截小后的模型分子保留其构象与生物活性构象一致,并作为后续Torsion Profile分析的起点。
QM扭转角分析(Torsion Profile)
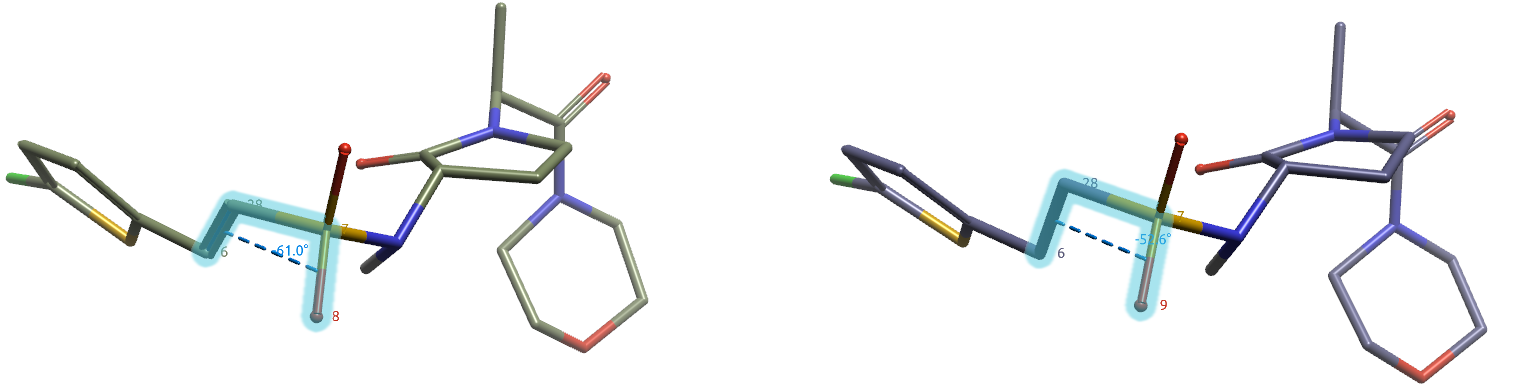
图2. 在QM Torsion Profile分析中定义5、6的两面角
以化合物5、6两面角(O=S)-(C=C)与(O=S)-(C–C)为例,如图2所示,用Flare QM的Torsion Profile对这两个两面角在BP86-D3BJ/DEF2-TZVP理论水平下进行扭转角分析,计算参数如下:
- Method: DFT
- DFT functional: BP86
- Use dispersion correction:Yes
- Basis set: def2-tzvp
- Convergence: Medium
- Max iterations: 500
- Number of rotomers: 72
- Degrees: 5.0°
- Max Threads: Auto
其中Degrees是根据Number of rotomers自动生成,无需主动设置,这里意味着以5.0°为步长进行两面角扫描。
结果与讨论
发现构象效应
虽然磺酰胺类抑制剂5、6(图1)在化学结构上仅存在一个单双键的差别,但是对Thrombin的Ki却存在20倍的差异,分别为367与17nM。为了理解化合物间的活性差异,Senger等人2用解释了5、6与Thrombin的X-ray复合物结构,PDB代码分别为2JH5、2JH6。将这两个结构下载到Flare,进行蛋白结构准备、共晶结构叠合,如视频1所示,可以发现5、6的各个原子重合的非常好,RMSD=0.2Å。最大的偏差来源自吗啉环上一个原子,距离为0.4Å。尤其注意到,具有结构差异的磺酰胺侧链则完全重合。
视频1. 化合物5与6在Thrombin结合位点里的叠合结果,PBD code分别为2JH5与2JH6
视频2分别呈现了化合物5、6与Thrombin各自的结合模式,我们可以进一步发现两者完全相同,蛋白的构象也没有差异。总的来说,从相互作用模式的角度讲,X-RAY共晶结构不能解释化合物5与6为何具有20倍的活性差异。
视频2. 化合物5、6与Thrombin的相互作用模式比较,PBD code分别为2JH5与2JH6
由于化合物5与6结构极其相似,两者之间不太可能存在与溶剂化、去溶剂化效应相关的能量差异,也不存在与蛋白结合相关的自由度损失差异。或者说,即使有,这两种差异不太可能带来20倍的活性差异。鉴于此,Senger等人2认为化合物5、6的活性差异仅可能是由于构象效应产生,并用QM方法对化合物5、6的两面角(O=S)-(C=C)与(O=S)-(C–C)进行了扭转角分析(Torsion profile)以验证假设。
化合物5、6的S-C两面角QM扭转角分析:降低构象张力能,增强活性
化合物5、6分别用简化的分子模型M1与M2代替,使用Flare QM对磺酰基S-C的两面角在BP86-D3BJ/DEF2-TZVP理论水平下进行扭转角分析,结果如图3、4所示。
需要注意的是,本文并没有完全采用Senger等人2的扭转角分析方法,而是进行了改良,因此存在几个方面的不同。首先,代表化合物5、6的模型分子不同了。在Senger等人的研究中,磺酰胺氮原子有单甲基取代,一方面这引入新的扭转角使得势能面扫描实际维度增加;另一方面氮原子的三角锥形翻转也使得势能面扫描增加更多的变化。为了保证势能面扫描充分且可靠,在文中的模型分子磺酰胺氮原子均没有甲基取代。第二,统一了扭转角分析策略。在Senger等人的研究中,M1与M2的扭转角分析策略不同:M1采用柔性扫描,而M2采用刚性扫描。我认为不同的策略可能导致势能面的可比性变差。因此在本文中所有的扭转角分析都采用柔性扫描。第三,使用了不同的理论化学模型。在Senger等人的研究中,使用了B3LYP/6-31G*,而在文本中使用了考虑色散效应的BP86-D3BJ/DEF2-TZVP。
如图3所示,当模型分子M1的(O=S)-(C=C)两面角为0或-115°时是有利的低能构象,但是这个构象不是化合物5的生物活性构象,PDB 2JH5里化合物5的该两面角为-61°,其能量比-0°附近的最低能构象高了不止3kcal/mol。
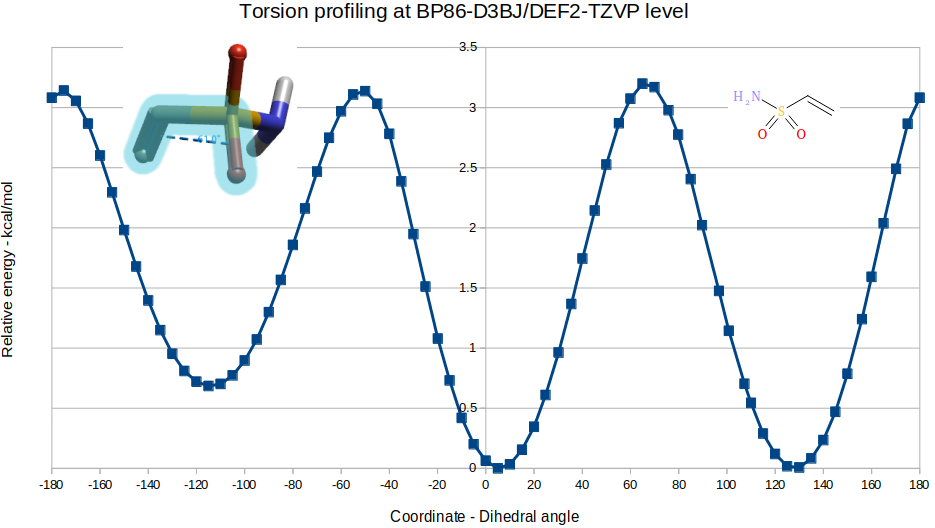
图3. 代表化合物5的模型分子M1及其在BP86-D3BJ/DEF2-TZVP理论水平下的Torsion profile结果
如图4所示,当模型分子M2的(O=S)-(C–C)两面角为-50°是低能构象,而这正是化合物6的结合构象,结合构象与最低能构象间不存在能量惩罚。
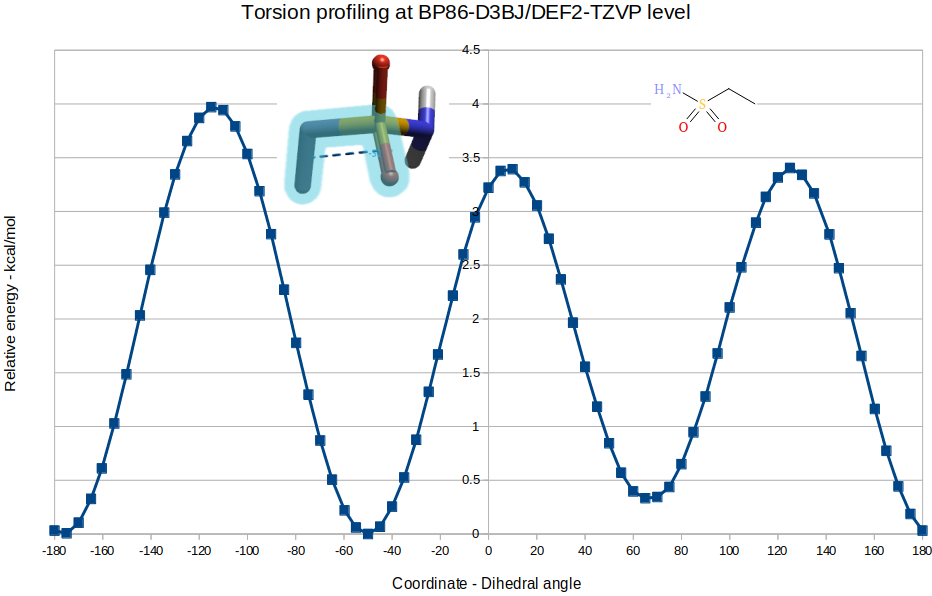
图4. 化合物6的模型分子M2在BP86-D3BJ/DEF2-TZVP理论水平下的Torsion profile结果
实际上,化合物6从结合构象出发往,两面角(O=S)-(C–C)沿着坐标的两边移动均存在陡峭的、大约3kcal/mol左右的能垒,这使得化合物6偏好于生物活性构象,最终导致比化合物5获得20倍的活性增强。
综上所述,化合物5、6与Thrombin具有完全一样的结合模式,结构生物学信息不能解释两者的20倍活性差异;而基于QM的两面角扫描分析结表明,与化合物6的生物活性构象相比,化合物5受到了不小于3kcal/mol的惩罚,这可用来支持化合物6与5的活性差异是因为构象效应带来的。
在SBD中使用构象效应指导先导化合物优化:化合物7的设计与验证
为了检验研究结果,Senger等人2试图通过结构修饰而不改变碳碳双键的键级来增加对Thrombin的活性。为了实现这一目标,必须稳定与靶标Thrombin形成复合物时的构象,也就是将化合物5的(O=S)-(C=C)两面角稳定在-60°左右。一种实现方法是将甲基连接到C=C双键噻吩偕位的碳原子上,从而设计得到化合物7(图1)。
用模型分子M3(图5)代替化合物7进行的扭转角分析,结果如图5所示,当两面角(O=S)-(C=C)为5、6生物活性构象的-50~-60°时,M3的相对能量约为0.9~1.14kcal/mol,这远低于模型分子M1的3 kcal/mol。
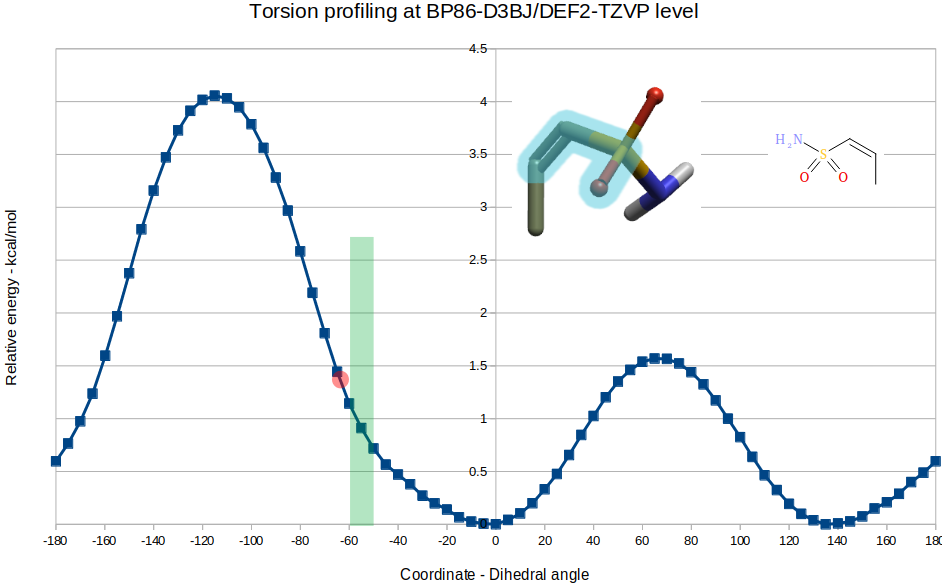
图5. 化合物7的模型分子M3在BP86-D3BJ/DEF2-TZVP理论水平下的Torsion profile结果。绿色区域:复合物结构PDB 2JH5、2JH6中生物活性构象两面角坐标区间(-50~-60°);红色点:复合物PDB 2JH0结构中化合物7的两面角与势能
因此,可以预期化合物7将比5具有更有效的Thrombin抑制活性,并假设7以与5和6相似的方式与Thrombin结合。非常令人鼓舞的是,后续的体外活性测试验证这个预测,据报道化合物7的Ki = 2 nM。
仅根据计算出的相对构象能量,发现7比6具有更有效的Thrombin抑制活性可能会令人惊讶。然而,必须考虑到7比5、6多了一个甲基,它有可能与活性位点中的残基发生有利地相互作用,与5相比可能会略微扰乱活性位点中配体的原子位置。为了排除构象效应之外的可能,Senger等人2解释了化合物7-Thrombin的X-Ray结构PDB 2JH0。结果表明,当与Thrombin结合时,7相对于5的RMSD为0.6 Å,两个分子之间等价原子之间的最大距离为1.0 Å,位于吡咯烷酮环上的一个碳原子。根据PDB 2JH0,化合物7的两面角(O=S)-(C=C)在生物活性构象时为-64°,根据扭转角分析,此时构象能大约为-1.4 kcal/mol,见图5红色圆点。这证实化合物7的活性提高仅与构象效应相关,构象效应可以作为定性指导广泛地用于基于结构的设计项目中。
关于2-乙烯基噻吩与2-乙基噻吩的扭转角张力能
如图6所示,化合物5、6、7的噻吩环与乙烯基、乙基的两面角张力能差异是活性差异的另一个来源。在生物活性构象时,化合物5、6、7的该两面角值分别为153、144、153°。
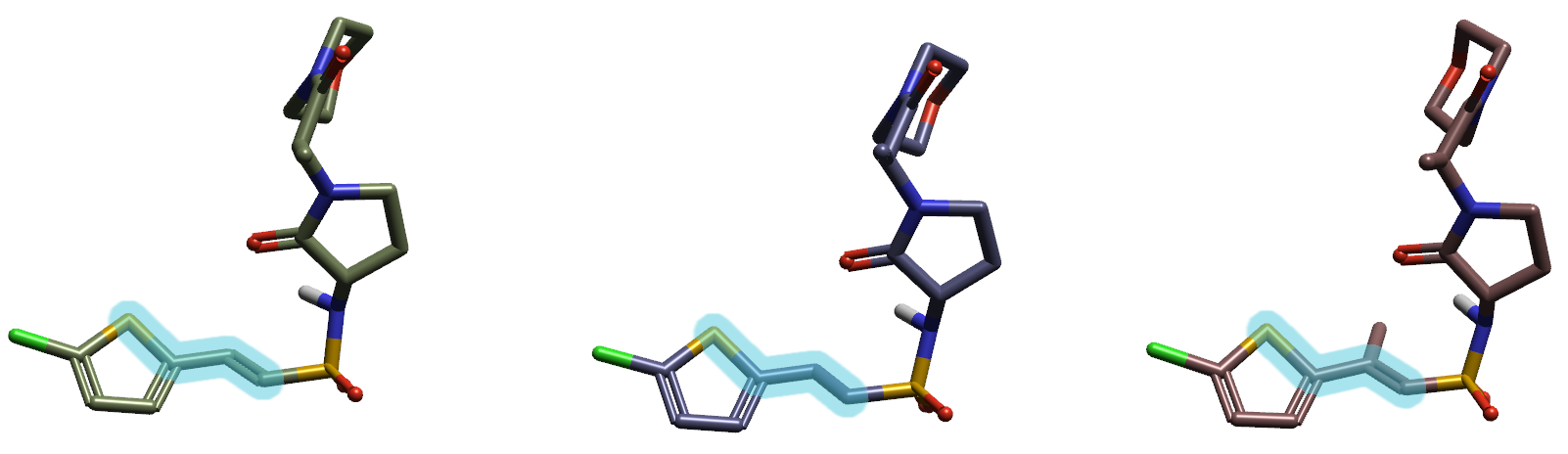
图6. 化合物5(左)、6(中)与7(右)的2-乙烯基噻吩与2-乙基噻吩的两面角
在PBE-D3BJ/DEF2-TZVP理论水平下对代表化合物5、6、7的模型分子进行扭转角分析,结果如图7所示,三者在生物活性构象时的张力能分别为~1.4、0.5、0.9 kcal/mol。
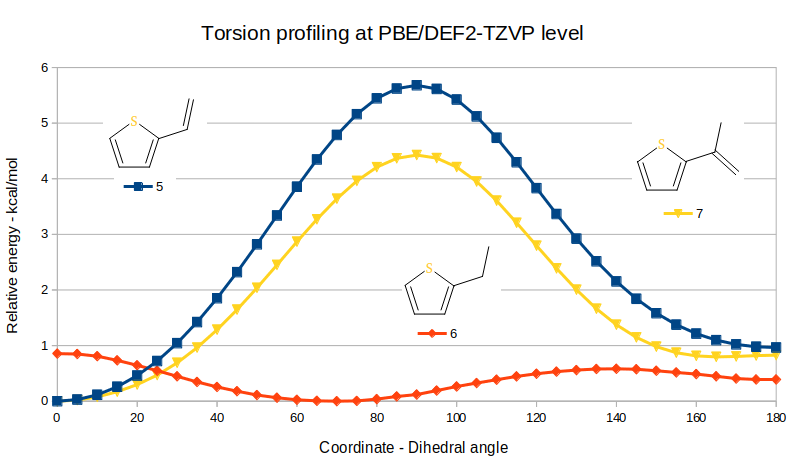
图7. 化合物5、6、7模型分子的2-乙烯基噻吩与2-乙基噻吩扭转角分析结果
显而易见,就噻吩-连接臂之间可旋转键的两面角而言,受到的张力能惩罚从高到低排序为:5,7与6。其中5受到的惩罚比6、7显著的高,这也与化合物5对Thormbin的活性低于6、7这个事实是一致的。
结论
在本文中,用Flare QM以回溯性地重现了文献报道的发现,表明构象效应在分子识别中起着重要作用。在基于结构的设计中,使用QM扭转角分析可以方便、可靠地考虑这类构象效应,并用于指导新分子的设计。
附件
输入文件下载:conformational_effect.tar.gz,解压之后的目录包含了PDB 2JH5、2JH6、2JH0的蛋白与配体结构,以及M1、M2与M3三个模型分子。
1 2 3 4 5 6 7 8 9 10 11 12 | conformational_effect ├── 2jh0_ligand_7.sdf ├── 2jh0_prot.pdb ├── 2jh5_ligand_5.sdf ├── 2jh5_prot.pdb ├── 2jh6_ligand_6.sdf ├── 2jh6_prot.pdb ├── M1.sdf ├── M2.sdf └── M3.sdf 0 directories, 9 files |
文献
- Murcko, Mark A. (1995). Computational Methods to Predict Binding Free Energy in Ligand-Receptor Complexes. Journal of Medicinal Chemistry, 38(26), 4953–4967. doi:10.1021/jm00026a001
- Senger, S. et al. (2007) “Sulfonamide-related conformational effects and their importance in structure-based design,” Bioorganic & Medicinal Chemistry Letters, 17(10), pp. 2931–2934. Available at: https://doi.org/10.1016/J.BMCL.2007.02.034.
- Flare V8. https://www.cresset-group.com/software/flare
- Flare QM. https://www.cresset-group.com/software/flare-qm


