
摘要:近年来,算力的可用性增加改善了配体-蛋白质相对结合自由能(relative binding free energy,RBFE)计算的可及性;然而,这些计算仍然需要大量的资源,这限制了它们的实用性。RBFE计算通常使用由转换坐标λ介导的一组热力学中间状态。优化λ提供了一种调整特定RBFE计算所需计算努力的方法。在此,我们介绍了自适应Lambda调度(daptive Lambda Scheduling,ALS),这是一种流畅的即时定制化的λ调度方法。我们的研究表明,它可以在保持预测性能的同时实现计算成本的显著降低。
原文:Midgley, S.D. et al. (2025) “Adaptive Lambda Scheduling: A Method for Computational Efficiency in Free Energy Perturbation Simulations,” Journal of Chemical Information and Modeling [Preprint]. Available at: https://doi.org/10.1021/acs.jcim.4c01668.
编译:肖高铿
通过炼金术式的原子级转化进行的相对结合自由能(Relative Binding Free Energy,RBFE)计算已经成为现代计算机辅助药物设计(CADD)的事实标准,用于获得同系物配体系列的可靠相对结合自由能1-3。近年来其受欢迎程度的增长部分得益于计算技术的进步,以及旨在增强RBFE工作流计算效率的算法的重大进展4-8。尽管有这些进展,对于许多用户和计算目的来说,RBFE计算仍然伴随着过高的计算成本,这继续推动了节省时间算法的开发。
目前,RBFE计算有几种现代方法,包括各种拓扑结构和副本(replica)表示。单副本方法涉及使用一个模拟路径来计算两个端点之间的自由能差值,而不是使用多个副本(副本交换)。单副本方法在理论上更为简单且资源消耗较少,但对中间状态的采样通常不如多副本方法严格。单拓扑方法涉及到通过操作力场中的价键、库仑和Lennard-Jones项,将起始分子(热力学状态0)中的原子逐渐变换为终点分子(热力学状态1)中的原子。当两个热力学端状态之间没有对应的原子时,会使用虚原子(Dummy atoms);然而,单拓扑方法允许的分子转化大小有限制。相反,双拓扑方法同时表示两个端状态,并调整力场项,使得一种物种被“淡化”为虚原子,而另一种则被“引入”到实际相互作用的原子中。这类方法的优点是可以处理更大的分子扰动,尽管它们更加复杂,通常比单拓扑方法更耗费计算资源。在本文中,我们专注于单复本、单拓扑的RBFE。对于理论方法的更详细比较,读者可以参考Mey及其同事的综述9。
单副本、单拓扑的RBFE(以下简称“RBFE”)计算的是不同热力学端状态(0, 1)之间的相对自由能差值,这两个端状态描述了两个不同的配体共享相同的蛋白质结合模式。这种能量差异是根据状态的概率来估算的,其中它们的相对系综势能可以通过分子动力学(MD)进行采样。由于端状态的相空间通常不重叠,因此仅考虑这些端状态通常无法实现有效的采样。相反,现代方法采用一系列沿炼金术转化坐标λ定义的中间状态。沿此坐标上的每个中间状态被称为一个λ窗口,而这些λ窗口的具体安排和总数则称为λ调度。
通过中间λ窗口对两个端状态的转化通常表示为一个重叠矩阵,该矩阵显示了沿坐标上每个窗口之间相空间重叠的程度。每个λ窗口在主对角线上表示,而任意两个相邻窗口之间的重叠则由直接位于主对角线上方(或下方)的次对角线表示,这条次对角线被称为超对角线。超对角线元素通常预期要达到某个最小值,以表明两个相邻的λ窗口足够接近,从而允许在中间状态下有合理的转化概率。这提供了一个方便的框架,用于表示和评估给定λ调度的适用性。作为说明性示例,读者可以阅读支持信息的说明性例子。
尽管λ调度对RBFE计算结果有显著影响,但大多数现代实现方法在进行炼金术转化时采用一组固定的λ窗口。非最优的λ调度可能导致λ窗口之间的重叠过多,这意味着不必要的采样和增加的计算成本;或者窗口间隔过宽,这可能导致最终自由能估算会产生的大误差。因此,找到一个最小可接受的λ窗口集是优化RBFE计算的一个重要因素。截至本文撰写时,已经开始出现一些通过特定转化的λ坐标调度展示出RBFE效率提升的方法10。特别值得注意的是Zhang等人10报告的优化相空间重叠(OPSO)方法。该方法使用短暂的“预热”计算来评估转化中的相空间重叠。虽然与本文介绍的方法存在概念上的相似性,但也存在若干重要差异。主要区别在于,OPSO方法通过对21个最大化的λ窗口集进行一轮“预热”计算来评估相空间重叠,而我们的方法从9个最小化的λ窗口集开始,采用迭代的方式,并且预计算时间显著更短(50皮秒对比OPSO的600皮秒)11。
我们在此介绍了一项RBFE计算机模拟研究,展示了新开发的自适应Lambda调度(ALS)协议在Cresset的Flare RBFE引擎内的应用,该引擎使用Sire分子模拟框架的SOMD应用程序12-13。通过在RBFE计算开始时使用一组初始低成本模拟获得的重叠矩阵,并基于此简单启发式算法计算定制的λ调度,我们结果表明取得了显著的时间节省。我们将ALS性能与等间距调度进行了比较,使用的数据集包括由Alibay等人14整理的分子片段数据集,这些数据来自各种先前的研究,以及由Wang等人15整理的P38抑制剂子集。完整的计算方法详见支持信息。
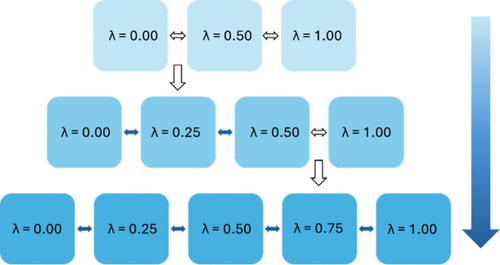
Flare的ALS协议总结在下图1a所示的示意图中。对于给定的转化(图1b),溶液中的游离配体(游离状态)会在第一次ALS迭代中初始化为101个可能等间距λ窗口中的9个。每个窗口运行50 ps 的分子动力学(MD),然后计算相空间重叠矩阵(图1b)16。识别出超对角线矩阵元素中小于阈值(默认为0.04)的元素,并在一个连续的低于阈值重叠区域(图1b中的红色方块)中插入一个新的λ窗口。当存在等长度的不良区域时,最低值的超对角线决定新窗口的插入位置。在两个端组之间的连续不良区域内插入新窗口时,该区域内所有窗口将被重新分布,以确保包括新窗口在内的所有窗口均匀分布。例如,在包含λ = 0.00, 0.06, 0.12的“不良区域”内插入一个新窗口并重新分配λ值后,更新的调度将是λ = 0.00, 0.04, 0.08, 0.12。对新窗口运行MD,并重新计算贡献/重叠,保留所有已运行窗口的信息。重要的是,之前执行的模拟结果(如第一次迭代中的λ = 0.00和0.12)得以保留,确保了计算资源的有效利用。用于计算ΔΔG值的多态Bennett接受率(MBAR)要求在状态λi处为所有其他状态λj计算能量17。考虑到101个可能的λ窗口使得在整个范围内计算能量成为可能,从而在无需重跑模拟或保留轨迹数据的情况下插入额外窗口。上述过程重复进行,直到没有低于阈值的超对角线矩阵元素,即预计算ALS组件已经收敛。一旦收敛,所有ALS的结果都将被丢弃,并使用收敛后的定制λ调度在给定转化的结合态和自由态中进行生产运行。
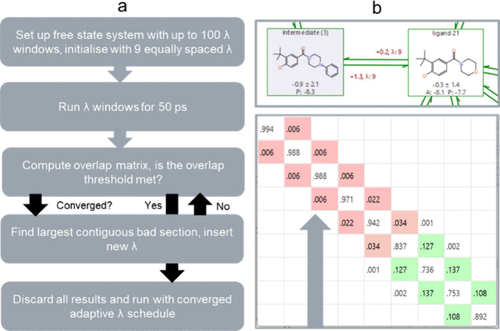
图1. a) ALS的工作流示意图;b) Flare RBFE图中展示的转化和重叠矩阵示例。
ALS的工作原理是基于重叠矩阵相对于模拟时间快速收敛,这意味着每个窗口仅需50皮秒的分子动力学(MD)模拟就足以估计任何给定转化的重叠矩阵。此外,重叠矩阵似乎主要由配体本身决定,而非其环境,这意味着可以从自由状态单独获得结合状态重叠矩阵的良好近似,而无需运行成本高得多的结合状态计算。这两个概念使我们可以使用短时间(50皮秒)的MD模拟来近似更严格的长时间(4-10纳秒)MD模拟,并且用自由状态的估计值作为自由状态和结合状态的良好近似。为了测试这些近似的有效性,我们评估了RBFE图中每个转换的近似设置和严格设置所给出的超对角线重叠矩阵元素。对于近似重叠矩阵超对角线的每个元素,根据是否满足重叠阈值(0.03、0.04或0.05),分配一个0或1的值。类似地,为严格的重叠矩阵生成了一个固定阈值0.03的相似值,从而为每对配体产生了一个近似和严格的二进制向量。通过混淆矩阵比较这两个向量,得到的统计信息使我们能够评估近似向量表示严格向量的能力。
补充信息中的表S2和表S3报告了通过上述混淆矩阵分析获得的统计结果,这些统计是使用Python的scikit-learn.metrics模块计算得出的18。50皮秒MD近似(表S2)产生了非常少的假阴性,这意味着我们很少会错过所需的λ窗口插入。相比之下,我们有较多的假阳性,意味着近似方法插入了多余的λ窗口。因此,该算法偏向于准确性和严格性而非速度。随着阈值的增加,假阴性的数量略有减少,但代价是假阳性的数量有所增加。在实际应用中,Flare的ALS结合了两种近似方法,使得50皮秒的自由状态被用作全长结合状态的代理。表S3展示了同时应用这两种近似的比较统计。从这里我们可以看到假阴性被最小化,意味着完整的(组合)近似方法保持了准确性,尽管可能无法获得真正最优的λ值集。总体而言,分析表明50皮秒MD和自由状态是全长MD和结合状态的良好近似,并且我们在享受ALS带来的效率提升的同时,不太可能牺牲科学的严谨性。
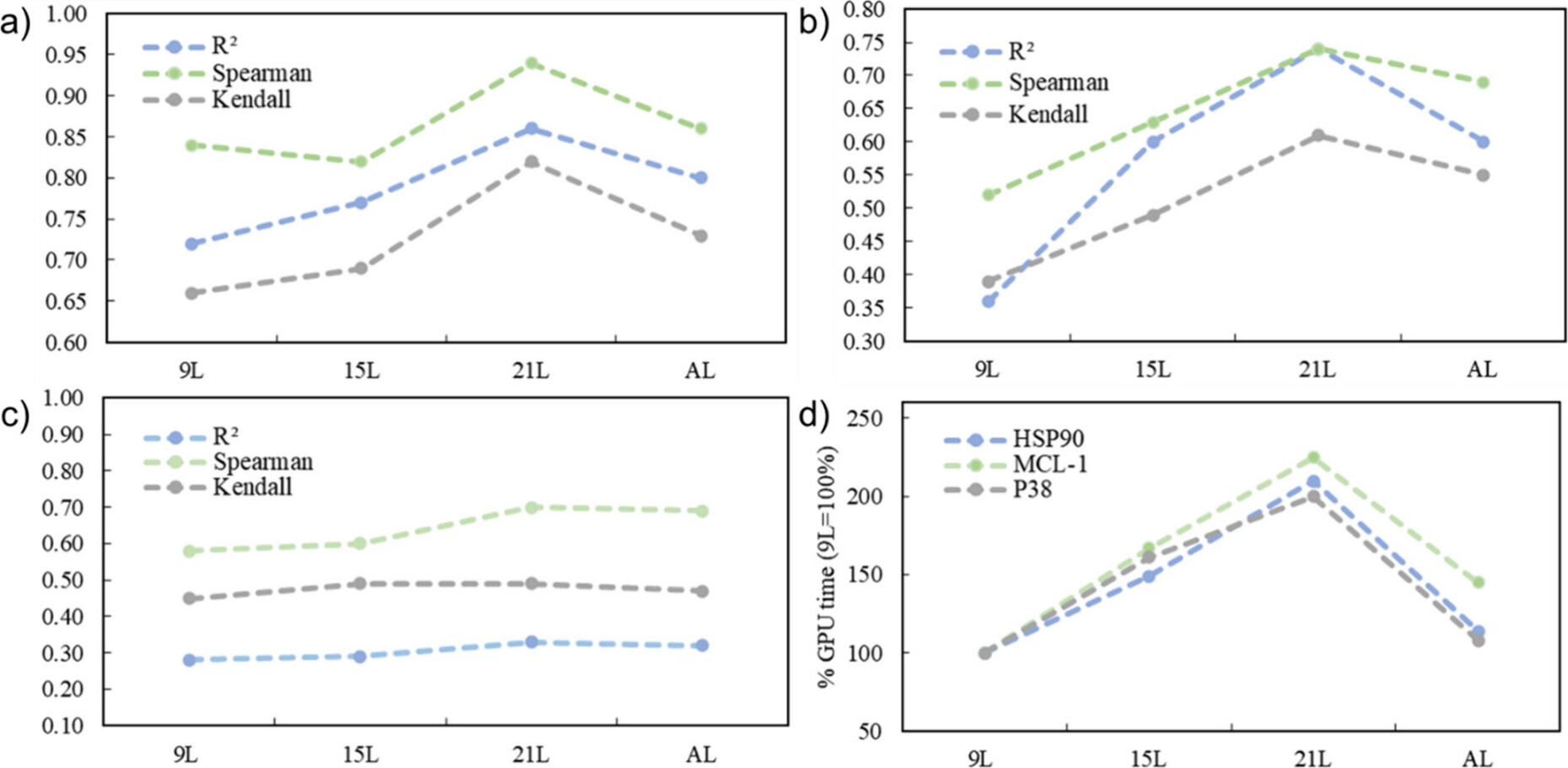
图2. 相关性统计量,分别为:a) HSP90,b) MCL-1,c) P38,以及 d) GPU时间。9L指的是由九个λ窗口组成的固定λ数组调度,15L = 15个λ窗口等,而AL指的是“自适应Lambda”,即完全应用了整个算法。
为了测试ALS的有效性,我们根据文献中15,19,20报告的实验值计算了结合亲和力预测值的R²、Spearman’s Rho和Kendall’s Tau。图2根据在RBFE图中使用的λ窗口数量报告了每个统计量。每个λ调度所需的相对GPU时间在图2d中报告。在HSP90和MCL-1算例中,正如预期的那样,使用更密集的λ数组时相关统计量通常有所改善,但代价是增加了GPU时间。ALS生成的统计结果与使用大量固定数量的λ窗口(15L-21L)相当,而其计算成本则与使用较小数量的λ数组(9L)相当。在P38算例上也表现出类似的趋势,尽管转向更密集的λ数组时相关统计量的改进不那么明显,这是由于在P38算例中配体-蛋白质体系的复杂性增加所致。ALS在所有三个研究系统中提供的相对趋势和效率增益是一致的(图2d)。为了检查该算法在一个更广泛的数据范围内的稳健性,我们将它应用于Wang等人(表1)的数据集15。如图2所示,该算法为这些数据集中超过97%的链接提供了良好的重叠矩阵,这表明在绝大多数情况下,在λ空间中获得了足够的采样。对于大多数链接,9个λ窗口已经足够,但在某些情况下最多添加了4个额外的λ窗口。
表1. Wang数据集中重叠矩阵超对角线元素小于0.03的链接的数量和比例15
| Target | No. of Links | Avg λ/link | Max λ/link | Poor Overlap | Bad overlap values |
|---|---|---|---|---|---|
| BACE | 54 | 9.0 | 10 | 0(0%) | |
| CDK2 | 22 | 9.3 | 12 | 0(0%) | |
| JNK1 | 27 | 9.4 | 11 | 0(0%) | |
| MCL1 | 63 | 9.2 | 12 | 2(3%) | 0.016,0.028 |
| P38 | 64 | 9.7 | 13 | 4(6%) | 0.009,0.019,0.020,0.027 |
| PTP1B | 39 | 9.5 | 12 | 2(5%) | 0.016,0.019 |
| Thrombin | 14 | 9.3 | 13 | 0(0%) | |
| TYK2 | 20 | 9.0 | 9 | 0(0%) | |
| Average | 38 | 9.3 | 11.5 | 1(2.6%) |
在所有RBFE设置中,我们避免为了控制目的而对λ调度或模拟长度进行临时调整,即使Flare的RBFE分析工具提示可能存在改进的空间。因此,在CADD情景下针对任何一个这些靶标进行研究时,通过迭代优化RBFE设置参数很可能会获得比报告中更高的性能统计结果。
总之,我们介绍了一种新颖的方法,用于提高计算机辅助药物设计(CADD)中RBFE计算的计算效率,并已在Cresset的Flare软件包中实现了这种方法。我们通过运行自由状态的短时间(50皮秒)模拟,为RBFE图中的每一对配体转化实时计算定制化的λ数组调度。我们已经证明,自由状态可以作为结合状态转化矩阵等效的有效代理,并且非常短的λ窗口模拟可以作为全长λ窗口模拟的有效代理,从而可以在最小的计算开销下构建自定义的λ调度。我们的结果表明,ALS有效地平衡了RBFE性能与计算效率,这对于希望在有限的计算资源上最大化RBFE通量的科学家来说非常具有吸引力。结果显示,使用ALS获得的R²通常与运行15-21个固定λ调度所得的结果相当。通过平均GPU时间和R²值,我们可以估计使用ALS相比15个固定λ调度大约可以减少37%的GPU时间,而相较于21个固定λ调度则大约可以减少50%的GPU时间。尽管这些节省的时间是基于有限数量的测试案例计算得出的,但我们估计ALS提供的节省时间通常会落在这个范围内。因此,我们认为特定转化的λ调度将成为现代CADD中RBFE的新标准的一部分。
数据获取
所有配体和蛋白质结构均可从公共开放访问数据库获取。蛋白质结构可以通过使用表S1中列出的PDB代码在蛋白质数据库(Protein Data Bank)中获得。来自各种先前工作的配体结构整理后,提供在GitHub仓库中的文件‘SMILES.csv’中,并附有用于生成统计报告的Python脚本。GitHub仓库可通过以下链接访问:https://github.com/cresset-group/adaptive-lambda-scheduling-article。计算机模拟是在Cresset的Flare软件包内进行的,该软件包是一款通过许可证获取的商业软件产品,详情请见https://www.cresset-group.com。关于Flare许可的更多信息可以在支持信息的第1部分找到。出于审查本文的目的,我们可以提供了完整的Flare许可证。对于有兴趣的读者,可以通过上述网址联系Cresset以获取试用许可证。
支持信息
支持信息可免费获取:https://pubs.acs.org/doi/10.1021/acs.jcim.4c01668
- 计算方法和结果,包括混淆矩阵统计、重叠矩阵、扰动图、链接和活性图(PDF格式):ci4c01668_si_001.pdf (1.24 MB)。
参考文献
略,请参考原文。
联系我们
想在自己的项目中亲自使用FLARE FEP,请联系我们获取免费的试用版;或者联系我在线演示功能;你还可以采购软件或委托研究与我们进行项目合作。此外,我们现在开始提供算力,按每分子计费,非常优惠。
- 电邮:info@molcalx.com
- 电话:020-38261356
原创文章,作者:小墨,如若转载,请注明出处:《自适应Lambda调度——一种提高自由能微扰模拟计算效率的方法》http://blog.molcalx.com.cn/2025/01/16/adaptive-lambda-scheduling.html


