
摘要:本文以DDR1抑制剂从苗头化合物到先导化合物的发现为例,演示了如何用Flare分子对接与AutoT&T的裁剪、移植策略相结合从分子对接结果里识别打分更优的片段、并自动将之自动裁剪、移植到起始化合物上,从而加速DDR1价抑制剂从苗头到先导的发现。
肖高铿/2021-11-29
1. 背景
DDR1 (Discoidin domain receptor 1)是一种胶原蛋白激活受体酪氨酸激酶,是一种由基质胶原激活的受体酪氨酸激酶,与癌症、纤维化和缺氧等疾病有关,因此而受到制药领域的关注。最近,Qiaoxin Tan等人[1]报道了综合利用深度学习实现、激酶选择性筛选、分子对接等技术实现从苗头化合物1(100nM时对DDR1的抑制率为48%)优化得到高选择性、高活性的DD1抑制剂先导化合物2(DDR1 IC50=10.6nM)。
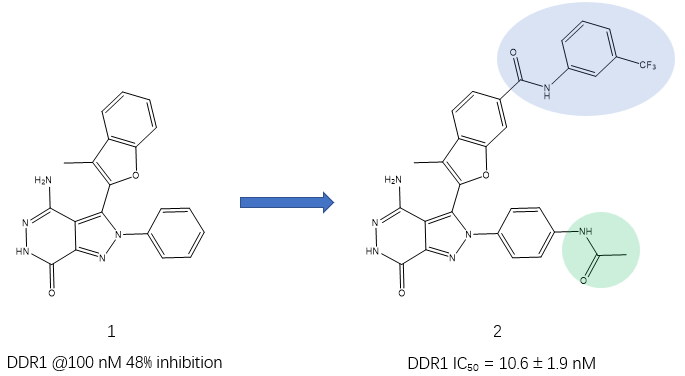
图1. DDR1抑制剂从苗头化合物1(DC1)到先导化合物2的优化
实际上,DDR1一直被作为模型靶标用来验证新设计方法的性能。比如Astex公司的Murray等人[2]在2015年就报道了采用片段设计策略,从苗头化合物3开始,通过分子连接得到DDR1/2高活性的先导化合物4(DDR1的IC50=1.5nM)。
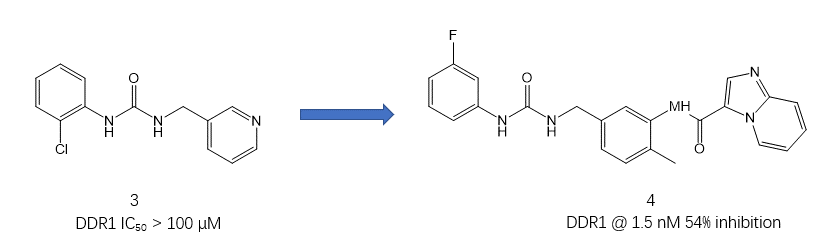
图2. DDR1抑制剂从苗头化合物3到先导化合物4的优化
2019年,Zhavoronkov等人[3]报道了首个采用深度生成模型结合药效团虚拟筛选技术发现的强活性DDR1/2抑制剂,其中代表性的化合物5对DDR1/2的IC50分别为10nM与234nM。

图3. DDR1/2抑制剂化合物5的化学结构
尽管深度生成模型能够生成具有所需性质的分子,但大多数性质并不包含诸如形状和药效团之类的3D信息。众所周知,源自3D形状和/或药效团的性质在药物设计过程中非常有用。Yoshimori等人[4,5]将深度分子生成框架REINENT与Ligandscout的3D药效团技术相结合,以药效团打分作为奖励函数进行增强学习生成满足DDR1抑制剂药效团模型的化合物。对其中9个化合物进行合成、测试之后发现,部分化合物6、7、8表现出良好的活性。
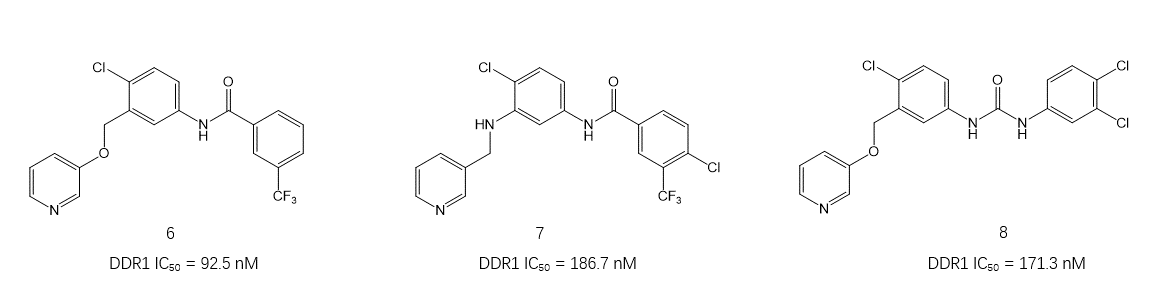
图4. DDR1抑制剂化合物6、7与8的化学结构
虽然深度学习生成模型目前引人注目,但并不代表其超越了传统方法,尤其在易用性。以Zhavoronkov等人3的文章为例,深度学习生成模型采集了大约30万化合物,需要进一步用药效团技术等多重过滤才富集到目标化合物,虚拟筛选在整个流程中起着重要作用。而使用生物等排体替换软件SPARK,仅需几分钟就可从现有化合物设计出高活性化合物5的衍生物[6]。
本文的主要目的是,采用比深度学习更容易操作、直观的分子对接技术与AutoT&T的裁剪移植技术相结合,来重现Qiaoxin Tan等人[1]报道的从化合物1到2的从苗头到先导的发现过程,最终为大家提供一种深度学习之外的技术方案选择,尤其当你想需要快速获得一些思路时。
2. 方法
2.1 蛋白结构的准备
将相关复合物晶体结构从蛋白质数据库(PDB)下载到Flare中,并使用来自Protein Prep工具小心地准备,以添加氢原子、优化氢键、消除原子冲突并给蛋白结构分配最佳质子化状态。任何截短的蛋白质链被封端作为蛋白质准备的一部分。使用COBALT多重比对工具在Flare中比对蛋白质序列,随后通过Cα的最小二乘拟合进行叠合。
2.2 小分子结构的准备
小分子以SMILES格式导入到Flare的配体表单(如图5所示),并采用基于规则的方式对化合物进行质子化状态处理,待用。
图5. 小分子导入到Flare的配体表单
2.3 分子对接
分子对接采用Flare Dock来完成,采用Template docking策略将化合物1以及13个ChEMBL化合物对接到PDB 5FDP的“干”(不考虑结晶水)结合位点里,采用如下参数:
- Calculation method:Very accurate but slow;
- Ligand to Dock: Select ligand(化合物1及13个ChEMBL化合物)
- Tempate Ligand:A 5WR 1001(参见结果部分,即PDB 5FDP的配体)
- GRID definition:ligand A 5WR 1001(参见结果部分,即PDB 5FDP的配体)
- Protein:5FDP
- Chains:A Chain
2.4 裁剪与移植:生成新的化合物
以化合物1位先导化合物,以共晶配体以及对接的13个化合物的pose为参比数据库,使用AutoT&T2[7,8,9]在结合位点从参比数据库中识别有利的片段,将之裁剪并移植到先导化合物1上,最终拼合出可能活动更好的化合物。
3. 结果
3.1 蛋白结构准备
人类DDR1的Uniprot获取号为Q08345,通过该获取号批量的从PDB上下载全部的蛋白结构、并进行序列比对,并将3D结构按Cα最小二乘拟合叠合到PDB 5FEP。整个过程采用Flare的python实现,代码如下:
上述的蛋白结构准备还将结合位点的配体提取到配体表单,将这些配体导出为ddr1-xtal-ligand.sdf备用。
现在我们可以通过3D视窗来观察蛋白结构准备的结果了。
3.2 配体结构的准备
将化合物1与13个从ChEMBL下载的已知抑制剂(见下表)导入到Flare配体表单。
1 2 3 4 5 6 7 8 9 10 11 12 13 | Fc1c(NC(=O)Nc2ccc(-c3c4c(N)n[nH]c4ccc3)cc2)cc(C)cc1 CHEMBL223360 FC(F)(F)c1cc(NC(=O)c2cc3c(C(C)CN(c4cncnc4)C3)cc2)cc(CN2CCN(C)CC2)c1 CHEMBL4087485 O=C(Nc1cc(C(C)C)cc(CN2CCN(C)CC2)c1)c1cc2c(C(C)CN(c3cncnc3)C2)cc1 CHEMBL4091837 Clc1cc(NC(=O)c2cc(C#CC3=Cn4nccc4N=C3)c(C)cc2)cc(CN2CCN(C)CC2)c1 CHEMBL2336010 FC(F)(F)c1cc(NC(=O)c2cc3c([C@@H](C)CN(c4cncnc4)C3)cc2)cc(CN2CCN(C)CCC2)c1 CHEMBL4097012 Clc1c(N)ncnc1C(=O)N[C@H](C)c1sc(C(=O)Nc2ncc(Cl)c(C(F)(F)F)c2)cn1 CHEMBL3348923 FC(F)(F)c1cc(NC(=O)c2cc(C#CC3=Cn4nccc4N=C3)c(CC)cc2)cc(CN2CCN(C)CC2)c1 CHEMBL2336015 FC(F)(F)c1cc(NC(=O)c2cc(c(CC)cc2)-c2cc(C)cc(-c3n4c(nc3)C=NC=C4)c2)ccc1 CHEMBL4555024 FC(F)(F)c1cc(NC(=O)c2cc(NCc3cnccc3)c(C)cc2)ccc1 CHEMBL4447700 FC(F)(F)c1cc(NC(=O)c2cc(NCc3cc(OC)cnc3)c(C)cc2)ccc1 CHEMBL4467377 O=C(Nc1ccc(C)cc1)c1cc(C#CC2=Cn3nccc3N=C2)c(C)cc1 CHEMBL2336035 O=C(NC(C)C)c1cc(C#CC2=Cn3nccc3N=C2)c(C)cc1 CHEMBL2336028 O=C(NC1CCCC1)c1cc(C#CC2=Cn3nccc3N=C2)c(C)cc1 CHEMBL2336032 |
3.3 分子对接
化合物1的对接到PDB 5FDP共晶结合位点产生了4个与铰链区结合的结合模式。在5FDP蛋白结合位点里,用MMFF94力场对每个pose进行了蛋白原子约束的能量优化,其中有一个pose与Qiaoxin Tan等人[1]报道的结合模式一致。如图6所示,化合物1与DDR1铰链区总共发生了三个氢键相互作用,其中与Met704发生两个氢键相互作用,化合物1的胺基与ASP702的羰基氧发生一个氢键相互作用。
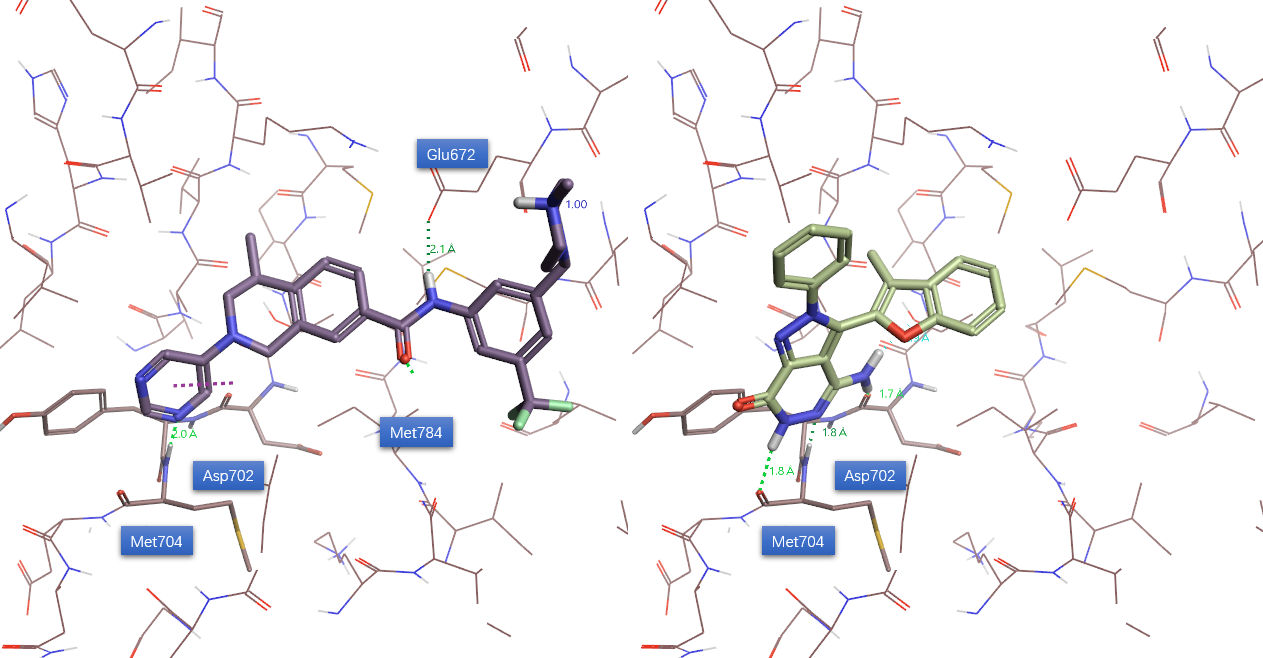
图6. 化合物1对接到PDB 5FDP共晶结合位点。左:PDB 5FDP共晶配体结合模式;右:化合物1预测得到的结合模式
比较化合物1与5FDP共晶配体的结合模式,如图7所示,正如Qiaoxin Tan等人[1]报道的那样:化合物1与5FDP的共晶配体匹配的相当好。尤其是5FDP共晶配体中与朝向别构位点酰胺连接的苯环几乎与化合物1的苯并呋喃的苯环完全重合,这直观地让人联想到将共晶配体N-(3-三氟甲基)-苯基甲酰基移植到化合物1苯上的设计。而这种策略正是Murray等人[2]采用的FBDD方法,已经证明有效且可靠。本文的主要目的是,利用AutoT&T的裁剪移植策略,自动从对接的配体里识别有利的别构位点结合片段,并移植到化合物1的苯环呋喃的苯环上。化合物1的另一个五员环氮上取代的苯环则朝向溶剂,在所有对接分子以及已知的共晶配体,尚未发现在该位置有相似的基团出现,因此本次演示并不包含该苯环的基团替换。
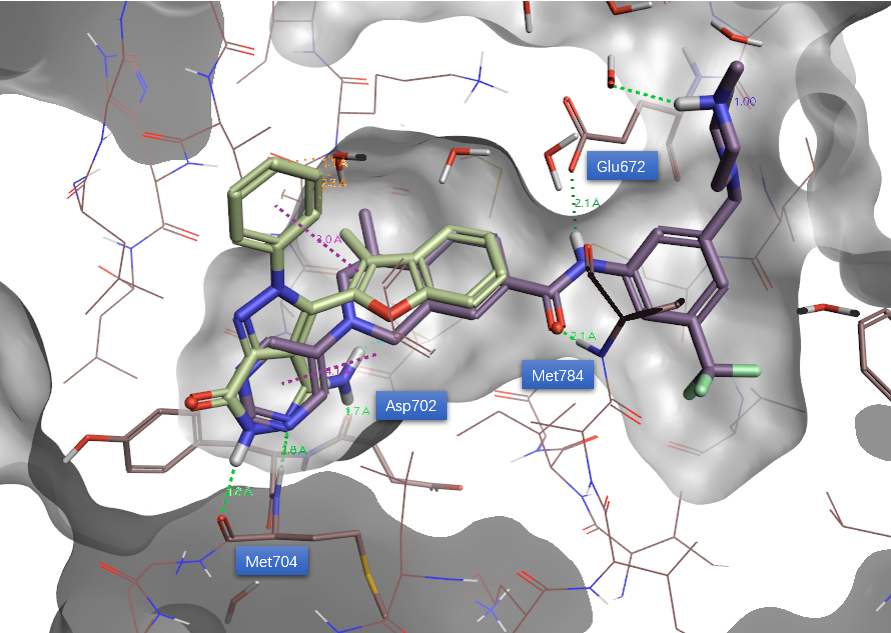
图7. 比较化合物1与PDB 5FDP共晶配体
13个从ChEMBL下载的化合物docking之后,每个化合物保存一个打分最佳的pose,保存为ddr1_chembl_dock.sdf备用;对应的蛋白结构删除水,仅保留A链,保存为5DFP_prot_dry.pdb。
3.4 AutoT&T:自动裁剪、移植的分子生长
在本次的裁剪、移植连接过程中,将被替换基团定义为图1化合物1苯并呋喃氧原子间位的C-H键,其原子序号分别为26、40。因为我们的目标是:希望将docking的pose以及从蛋白结合位点里提取到的配体里面适合替换H40片段裁剪下来,移植到C26上,所以我们将C26-H40单键定义为优化位点,以便从这个C12位置上往别构位点方向上生长出新的片段。AutoT&T的LinkLeadOpt可以帮助我们实现这个片段裁剪、连接的策略。
在进行分子生长之前,先将从共晶结构得到的配体与docking得到的配体合并:
1 | cat ddr1_xtal_ligand.sdf ddr1_chembl_dock.sdf >> ddr1_inhibitor_merge.sdf |
接着用AutoT&T2的LinkLeadOpt来进行分子生长:
1 2 3 4 5 6 | LinkLeadOpt -l DC1_dock.sdf \ -vs ddr1_inhibitor_merge.sdf \ -p 5fdp_prot_dry.pdb \ -out ddr1_linked.sdf \ -c '(26,40)' \ -ih |
其中,参数-l读入先导化合物(本例中为化合物1对接后的结果:DC1_dock.sdf); 参数-vs读入参考分子库,也就是在3.1节导出的共晶配体与3.3节得到抑制剂对接pose合并后的化合物; 参数-p读入3.3对接计算用的蛋白结构,删除了水仅保留A链;参数-c指定了优化位点,若剪切下来的片段在打分上优于氢原子,则进行片段替换(即分子生长);参数-out用来指定输出文件的名称;-ih表示C-H参与替换的键匹配。
AutoT&T总共生成了7个化合物,其中有三个结构相同(三个分别来自PDB 5FDP、5FDX的片段与来自CHEMBL4087487的片段结构相同),因此聚类总共得到5个化合物,如图8所示。
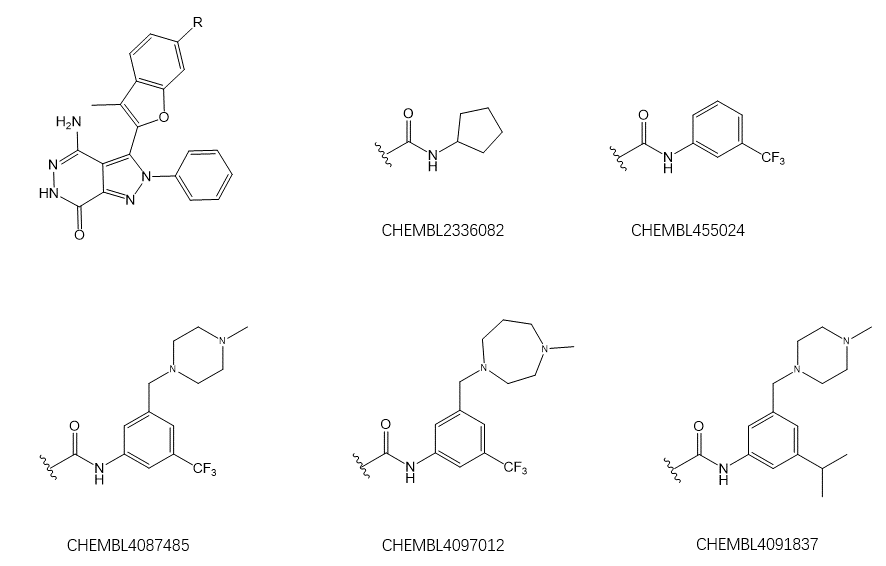
图8. AutoT&T生成的5个化合物
其中来源自CHEMBL455024的N-(3-三氟甲基)-苯基甲酰基正是Qiaoxin Tan等人[1]报道的化合物2的片段。该化合物与DDR1的结合模式如图9所示,保留了DC1与铰链区残基Met704两个氢键、Asp702一个氢键的相互作用;此外新引入的N-(3-三氟甲基)-苯基甲酰基则占据了别构位点。其中酰胺羰基氧作为受体与Met784的NH发生氢键相互作用,酰胺NH作为供体与Glu672末端的羧基发生氢键相互作用。AutoT&T确实从对接获得配体结合模式中识别到优势片段并将之移植到预期的优化位点上。
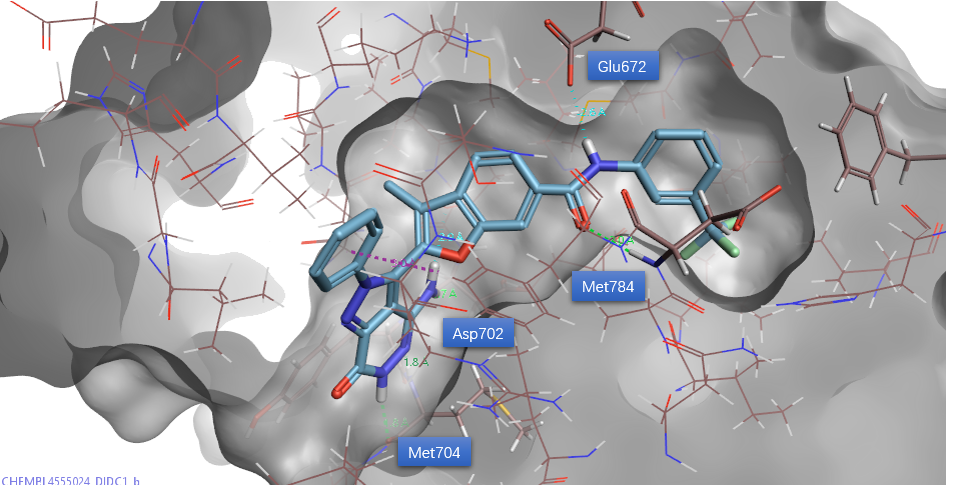
图9. DC1与CHEMBL455024组合出的化合物的结合模式
4. 小结
我们采用一个python工作流从PDB数据库上批量下载了DDR1蛋白结构,并进行了标准的结构准备、序列比对、蛋白叠合、配体提取。我们还从ChEMBL上下载了13个已知的DDR1抑制剂,并用Flare将DC1与下载的化合物对接到已经准备好的PDN 5FDP的共晶结合位点里。
我们以DC1(化合物1)为起点,以其苯并呋喃氧间位的C26-H40为优化位点,AutoT&T自动的从docking到结合位点的已知抑制剂上识别有利的片段,并将之裁剪、移植到起点化合物的C26-H40上生成新的5个化合物。其中来源自CHEMBL455024的N-(3-三氟甲基)-苯基甲酰基正是深度学习方法获得关键结构片段。
与深度学习结构生成相比,AutoT&T不需要进行生成模型训练,仅需要分子对接结构作为片段来源进行裁剪与移植。分子对接是常规药物发现工作流程的一部分,可以来源于已有的虚拟筛选结果,而不必一定是专门为裁剪、移植而进行虚拟筛选。相对于虚拟筛选通常仅选择少量化合物进行测试而言,这是一种对虚拟筛选结果进行充分再利用的一种高效方式,也是高效的药物化学分子设计策略,为您的项目快速地产生新的思路。
5. 接下来做些什么
5.1 结合构象优化
需要注意的是,AutoT&T生成的ddr1_linked.sdf的结构还需要进一步在结合位点里进行能量优化,以便移植过来的片段与原有DC1之间的键在键长与键角上是合理。在Flare里很简单,在配体表单里选中要优化的配体,点击Minimize, 选择对应的蛋白结构就可以开始优化配体了。
5.2 构象稳定性评估
不稳定的结合构象或高能构象是分子对接虚拟筛选假阳性的主要原因之一。为了保证生成分子的结合构象是稳定的,需要进一步进行构象稳定性评估。如何评估构象稳定性,在之前的博客里有很多讨论,请在我们的博客网站里搜索“构象稳定性”与“Torsion profile”,这里就不再叙述。
6. 附件
数据集下载:ddr1_5fdp.tar.gz
7. 文献
- Tan, X.; Li, C.; Yang, R.; Zhao, S.; Li, F.; Li, X.; Chen, L.; Wan, X.; Liu, X.; Yang, T.; et al. Discovery of Pyrazolo[3,4- d ]Pyridazinone Derivatives as Selective DDR1 Inhibitors via Deep Learning Based Design, Synthesis, and Biological Evaluation. J. Med. Chem. 2021. https://doi.org/10.1021/acs.jmedchem.1c01205.
- Murray, C. W.; Berdini, V.; Buck, I. M.; Carr, M. E.; Cleasby, A.; Coyle, J. E.; Curry, J. E.; Day, J. E. H. H.; Day, P. J.; Hearn, K.; et al. Fragment-Based Discovery of Potent and Selective DDR1/2 Inhibitors. ACS Med. Chem. Lett. 2015, 6 (7), 798–803. https://doi.org/10.1021/acsmedchemlett.5b00143.
- Zhavoronkov, A.; Ivanenkov, Y. A.; Aliper, A.; Veselov, M. S.; Aladinskiy, V. A.; Aladinskaya, A. V; Terentiev, V. A.; Polykovskiy, D. A.; Kuznetsov, M. D.; Asadulaev, A.; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019. https://doi.org/10.1038/s41587-019-0224-x.
- Yoshimori, A.; Asawa, Y.; Kawasaki, E.; Tasaka, T.; Matsuda, S.; Sekikawa, T.; Tanabe, S.; Neya, M.; Natsugari, H.; Kanai, C. Design and Synthesis of DDR1 Inhibitors with a Desired Pharmacophore Using Deep Generative Models. ChemMedChem 2021, 16 (6), 955–958. https://doi.org/10.1002/cmdc.202000786.
- 肖高铿.使用深度生成模型设计具有所需药效团的DDR1抑制剂.墨灵格的博客.2021-07-03. http://blog.molcalx.com.cn/2021/07/04/ddr1-inhibitor-design-using-deep-generative-models.html
- 肖高铿. 用SPARK基团替换策略重现深度学习DDR1抑制剂的发现.墨灵格的博客.2019-09-05.http://blog.molcalx.com.cn/2019/09/05/spark-ddr1.html
- Li, Y.; Zhao, Y.; Liu, Z.; Wang, R. Automatic Tailoring and Transplanting: A Practical Method That Makes Virtual Screening More Useful. J. Chem. Inf. Model. 2011, 51 (6), 1474–1491. DOI:10.1021/ci200036m
- Li, Y.; Zhao, Z.; Liu, Z.; Su, M.; Wang, R. AutoT&T v.2: An Efficient and Versatile Tool for Lead Structure Generation and Optimization. J. Chem. Inf. Model. 2016, 56 (2), 435–453.DOI:10.1021/acs.jcim.5b00691
- AutoT&T 2.0-高效的药物分子从头设计软件.墨灵格的博客.2018-08-23. http://blog.molcalx.com.cn/2018/08/23/atnt2.html


