
摘要:本文以PD-1/PD-L1相互作用抑制剂BMS-202为例,用Flare/GIST与Ligandscout/Apo site grid分析了其与PD-L1的共晶结构结合位点,识别出可替换的HOH350以及子口袋引入新相互作用的假设;利用SPARK将Flare与Ligandscout的假设转化为虚拟筛选查询条件与约束条件,进行了水分子替换虚拟筛选,重现了文献报道的引入3-腈基苄基的先导化合物优化过程。这为子口袋引入新的相互作用提供了一个高效的通用计算方法。
肖高铿/2022-07-07
1. 前言
用单克隆抗体阻断PD-1/PD-L1免疫检查点通路是癌症治疗领域的重大进展。基于抗体的免疫疗法具有许多缺点,比如抗体的高成本、有限的半衰期和免疫原性。此前,由于该通路的结构生物信息学不完整,能够克服这些缺点的小分子PD-1/PD-L1抑制剂的开发进展缓慢。
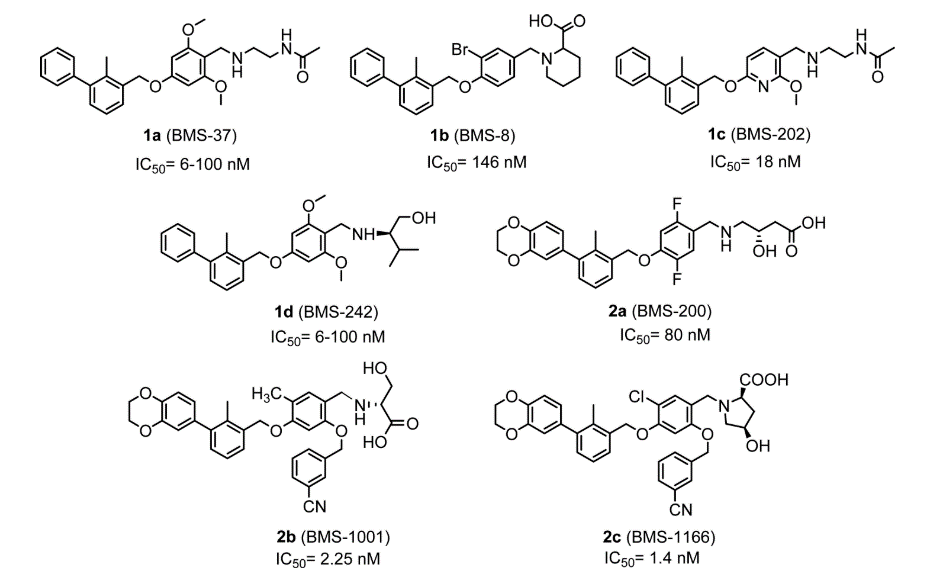
图1. 几个已知的PD-1/PD-L1相互作用抑制剂
在Bristol-Myers Squibb首次披露系列非肽类PD-1/PD-L1相互作用抑制剂(图1)之后,Zak等人[1-3]进行的结构生物学研究揭开了部分化合物与PD-L1的相互模式,这为理解这些化合物的SAR提供了坚实的基础。
图2. 化合物1c(BMS202)与PD-L1的共晶结构(PDB 5J89)的相互结合模式
观察化合物1c(BMS-202)与PD-L1共晶结构(PDB 5J89)所揭示的蛋白-配体相互作用(图2),可以发现在母核吡啶的甲氧基所在处有个子口袋,在甲基西边6.8Å处有一个水(HOH350A)与ARG125A的酰胺-NH发生氢键相互作用(见图3-左图)。将化合物2c(BMS-1166)与PD-L1共晶结构(PDB 6R3K)叠合PDB 5J89上,如图3-右图所示,会发现化合物2c的3-腈基苄基从1c的甲基出发往子口袋的HOH350A方向延伸、其中腈基替换了HOH350A并作为氢键受体与ARG125A的-NH发生氢键相互作用;而苄基一方面与TYR123A发生芳香-芳香的pi-pi相互作用,另一方面与LYS124A发生阳离子-pi相互作用,在3-腈基苄基片段上这些新引入的相互作为1c到2c的先导化合物优化贡献了有利的蛋白-配体相互作用。

图3. 将化合物2c与PD-L1的共晶结构(PDB 6R3K)叠合到化合物1c与PD-L1的共晶结构(PDB 5J89)上。左图:PDB 5J89中未被1c占据的结合子口袋;右图:PDB 6R3K共晶配体(化合物2c)叠合到PDB 5J89的结合口袋上。
我感兴趣的问题是:如果只有化合物1c与PD-L1的共晶结构(PDB 5J89),能否利用基于结构的药物设计方法得到在吡啶母核的甲氧基侧链上引入3-腈基苄基来实现先导化合物优化的方案?此外,高活性化合物2b与2c由于分子量过大有别于传统的类药分子,用传统的对类药、类先导化合物库进行虚拟筛选的策略命中不了该类化合物,通过对结合位点分析引入新相互作用是优化该类分子的重要手段。因此,本文尝试使用Flare/GIST与LigandScout/Apo Site Grid等基于结构的方法对PDB 5J89结合位点进行分析以获得针对子口袋进行结构优化的假设,然后通过SPARK进行侧链虚拟筛选生成满足假设的化合物。结果发现,在新生成的化合物中包含了文献报道的设计。这说明,我们基于结构的设计策略可以可靠地在子口袋引入新的相互作用实现高效的先导化合物优化。
2. 结果
2.1 用GIST分析结合位点Apo结构水的热力学性质
首先用Flare的GIST对PDB 5J89的apo口袋进行水的热力学性质分析。在PDB 5J89的结合口袋里有三个水HOH301A、HOH316A与HOH350A,对apo结构的GIST分析结果表明,这个三个水位置计算的水密度大于4,如下图4所示。

图4. GIST分析共晶结构PDB 5J89结合位点水的密度大于4的区域。
图5显示了GIST方法对Apo结合口袋分析的ΔG等值图,其中红色区块为ΔG大于0.5kcal/mol的区域,而绿色区块为ΔG小于-0.5kcal/mol的区域。结果表明,结合位点基本被红色的区块填充,而没有绿色区块出现,这说明该结合位点具有良好的成药性:因为红色等值图意味这些水可被替换,有可能设计出小分子替换这些水而与蛋白结合。HOH301A、HOH316A与HOH350A三个水所在位置的ΔG均大于2kcal/mol,这是相当高能的水,而且这三个水还是GIST分析的水高密区,对这些高能量、高密度水的替换或置换可能带来结合亲和力的提高。

图5. GIST分析共晶结构PDB 5J89结合位点ΔG大于0.5kcal/mol(红色)与ΔG小于0.5kcal/mol(绿色)区域。
进一步分析apo口袋水的焓变,如图6所示,HOH301A、HOH316A与HOH350A三个水所在位置的ΔH均小于-2kcal/mol,这主要是因为HOH350A与ARG125A的酰胺NH发生氢键相互作用;HOH301A与HOH316A不仅与蛋白残基发生氢键相互作用,还同时与配体、其它结合水发生氢键相互作用,这些相互作用形成了稳定的氢键网络,因此它们所在位置的ΔH相当小(小于-2.0kcal/mol)。需要注意的是,其中HOH350A处的绿色等值图非常小,以至于在图6里呈现的并不清楚。

图6. GIST分析共晶结构PDB 5J89结合位点ΔH大于2kcal/mol(红色)与ΔH小于2kcal/mol(绿色)区域。
对PDB 5J89的apo口袋进行的水GIST分析得到热力学性质可总结为图7,其中HOH301A、HOH316A与HOH350A三个水所在位置的ΔG均大于2kcal/mol,而ΔH均小于-2kcal/mol。这说明,如果对这个三个位置的水分子进行替换的话,新引入的取代应基维持与蛋白残基的相互作用、以及维持氢键网络,如此则可能有助于提高化合物的活性。

图7. 共晶结构PDB 5J89结合位点3个apo结构高能的水。
进一步的分析表明,HOH301A与HOH316A的稳定化自由能ΔGstablization小于0(将在另一个博客中介绍计算过程),它们对蛋白-配体复合物结构起到稳定化的作用。这意味着对HOH301A与HOH316A还可以采取保留作为蛋白一部分的策略。
2.2 Apo site grid分析发现芳香-芳香相互作用
鉴于PDB 5J89的吡啶环甲氧基与HOH350A处于一个明确的子口袋里,而如前所述,HOH350是一个高能、高密度的水,从甲基处出发引入取代基替换HOH350A水自然而然成为结构优化的一个选择。因此有必要进一步了解这个子口袋的特性,以了解在该子口袋存在的其它有利相互作用形式以便在后续的设计加以考虑。Ligandscout的apo site grid可以用来分析蛋白结合位点并给出可能的有利药效团相互作用特征。
在Apo site grid分析之前,将HOH350A与HOH316A移到到配体的一边,而将HOH301A保留在蛋白一边,删除其它水。然后用基于结构的Apo site grid方法生成药效团模型,结果如图8所示。HOH350A做为氢键受体与蛋白ARG125A的酰胺NH发生氢键相互作用;HOH316A桥接了一个氢键网络,与ASP122A、TYR123A以及LYS126发生氢键相互作用;配体吡啶环甲氧基与HOH350之间距离为6.8Å。Apo site grid分析结果表明:在甲基与HOH350之间有个芳香药效团特征与TYR122A发生芳香-芳香相互作用。
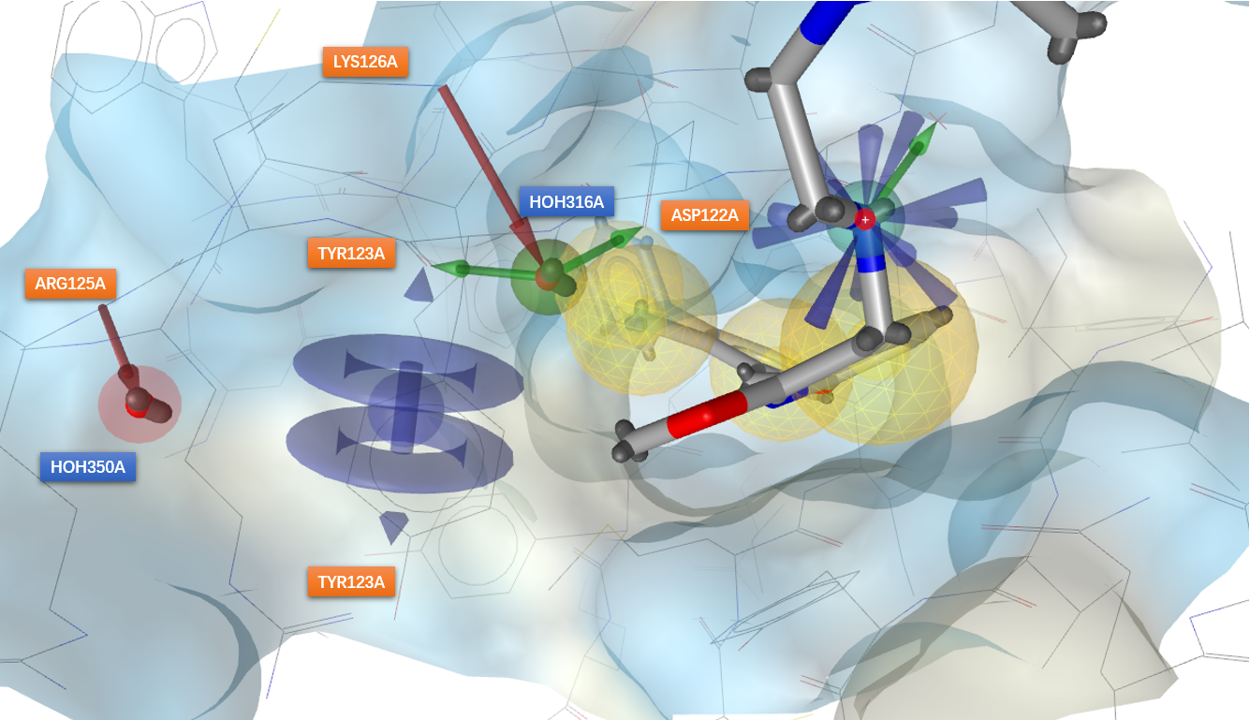
图8. 用LigandScout/Apo site grid对共晶结构PDB 5J89结合位点进行药效分析的结果
如图8所示,我们还可以发现HOH316A与配体正电中心相连C之间的距离很近,仅3.6Å,相当于两个键的距离。HOH316A作为配体的一部分,发挥着既是氢键受体也是受体的功能,这很容易让人联想到用羟基代替该水,但这不是本文要讨论的重点。
2.3 用SPARK进行水分子替换的侧链虚拟筛选
根据上述2.1与2.2小节的分析我们可以得到在子口袋引入新相互作用的假设,如图9所示,包含两个必要药效团特征:1)一个氢键受体以便替换HOH350A与ARG125A发生氢键相互作用;2)一个芳香中心以便与TYR123A发生pi-pi相互作用,有可能的话同时与LYS124A发生阳离子-pi相互作用。
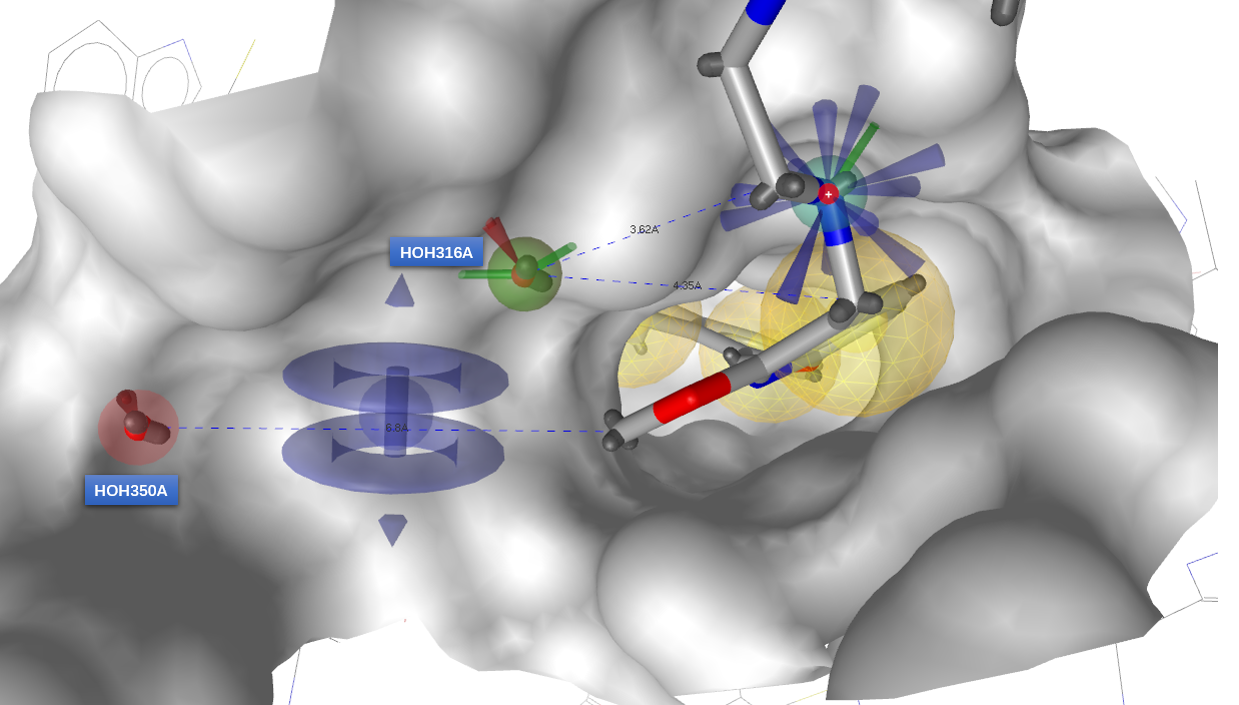
图9. 在子口袋引入新相互作用的假设
根据该假设,用SPARK的水分子替换策略对甲基与HOH350A进行替换的虚拟筛选,在500个打分最高的结果中包含了43个含腈基取代的苯环或吡啶环的化合物,如图10所示。

图10. 43个含腈基取代苯环或吡啶环的结果
具体地讲,如图11所示,文献报道的、高活性的3-腈基苄基片段[2]与3-腈基吡啶-5-亚甲基片段[5]及其衍生被SPARK命中,也被相关的复合物结构PDB 6R3K与6RPG在原子水平上所证实,SPARK生成的化合物可用分子对接方法进行下一轮的虚拟筛而进一步富集出这些化合物。
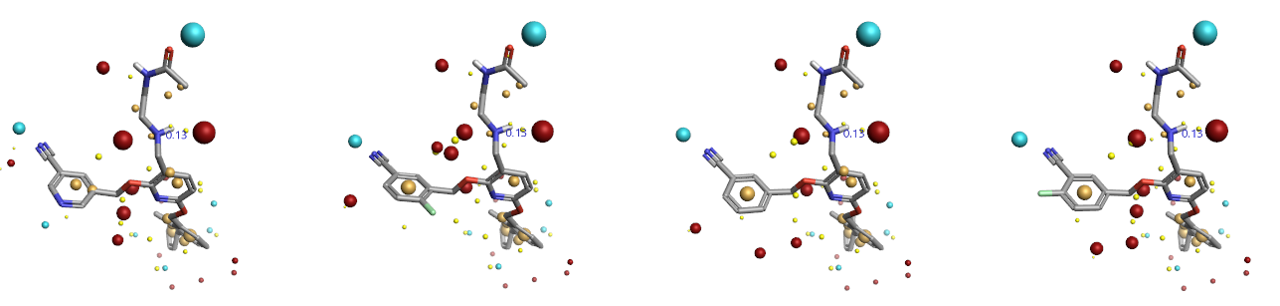
图11. 文献报道的两个侧链及其衍生物
总的来说,根据Flare/GIST与Ligandscout/apo site grid基于结构分析得到的假设进行的SPARK虚拟筛选可以命中被文献报道的高活性化合物,这进一步证实了该方法的可靠性。
3. 材料与方法
3.1 蛋白结构准备
将化合物1c与2c与PD-L1的共晶结构(PDB 5J89与6R3K)从蛋白质数据库下载到Flare V6[6]中,并使用其中的Protein Prep工具小心地准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构分配最佳质子化状态。任何截短的蛋白质链被封端作为蛋白质准备的一部分。将1c与2c结构从分别从准备好的共晶结构中提取出来。
3.2 GIST分析
了解蛋白活性位点内水分子的行为是药物设计的一个非常重要的方面。在蛋白质-配体复合物中,了解桥连水分子的稳定性是决定药物设计策略的关键。如果水分子不是特别稳定,可以设计配体以取代它,并与蛋白质活性位点直接相互作用:这通常会导致配体生物活性的增加。
GIST是一种水热力学性质分析方法[7],用于评估结合口袋的水合作用,并通过在分子动力学运行结束时对显式溶剂分布采样来计算相关的水热力学性质。GIST分析的结果显示为活性位点水合作用的三维等值面映射“happy”(绿色,与负∆G相关)和“unhappy”(红色,与正∆G相关)区域。unhappy区域映射的是蛋白质活性位点内更可能的可成药区域。happy区域映射的是蛋白结合位点内较低可能成药的区域,在那里水分子更稳定,因此更难置换。
在本文中,用Flare V6分别对结合位点的Apo结构(不包含配体、金属离子与结晶水)进行了GIST分析,使用了如下条件:
- Calculation method: Normal
- Ligand: None
- Grid spacing:0.5 Å
- Grid Definition:Ligand
- Chains: A
- Simulation length: 20ns
- Solvent Model: explicit TIP4Pew Water
3.3 Ligandscout/Apo site grid分析
将PDB 5J89共晶结构下载里Ligandscout 4.4.8[8],选择B链的配体来定义结合位点,将HOH350A与HOH316A移到配体一边,将除了HOH301A之外的其它水删除,生成基于结构的药效团模型。然后在定义的结合位点里,用Create Apo site grid生成药效团模型,在默认的阈值下仅分析药效团特征AR分布,如图12所示。

图12. PDB 5J89的Ligandscout/Apo site grid分析
如果将Ar相互作用特征阈值设置为1.0,则发现芳香相互作用特征主要分布在HOH350A与甲氧基之间,这为后续SPARK水分子替换采用芳香相互作用约束条件提供了有力的支持。
3.3 SPARK水分子替换虚拟筛选
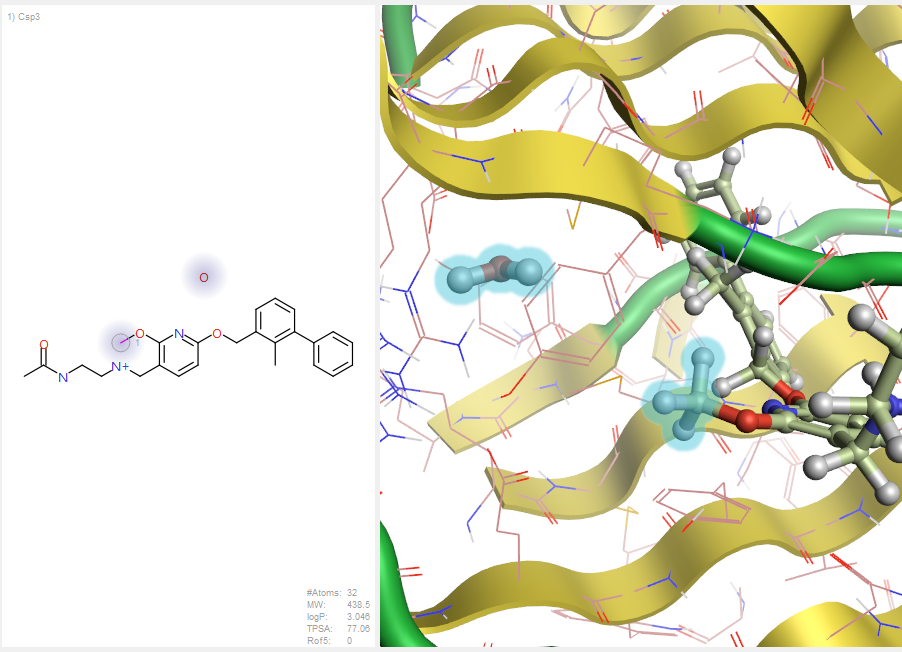
图13. SPARK水分子替换的query
用SPARK V10.6.0的水分子替换策略对甲基与HOH350A进行虚拟筛选,如图13所示,以PDB 5J89的HOH350A水为被替换水,设置共晶配体的甲基为被替换基团。并在HOH350设置氢键受体的约束条件,如图14所示,以确保满足氢键受体的假设。
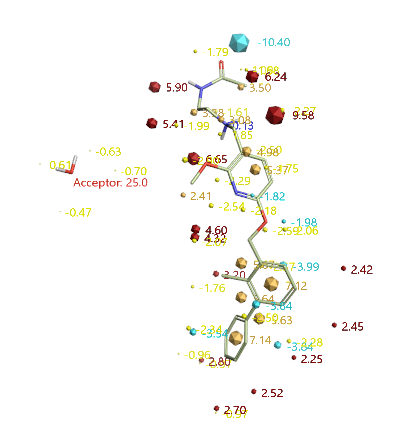
图14. SPARK水分子替换query以及HOH350的氢键受体药效团约束
出于演示的目的,本文仅选择了ChEMBL common, Commercial Verycommon与common等3个常见的片段库进行虚拟筛选,如图15所示。更多的数据库信息请参阅文献4。
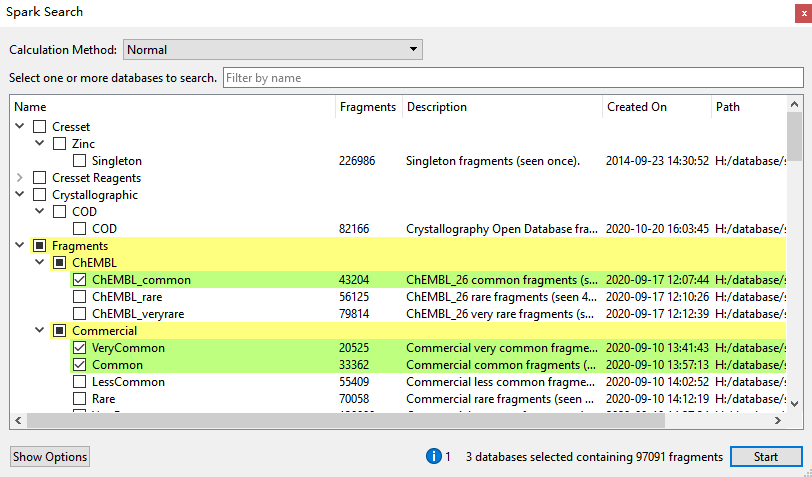
图15. SPARK水分子替换虚拟筛选的数据库
鉴于我们的假设包含两个药效团特征,因此还使用了如图16的过滤器来作为虚拟筛选的条件,以确保搜索到的化合物更加满足我们预设的条件。
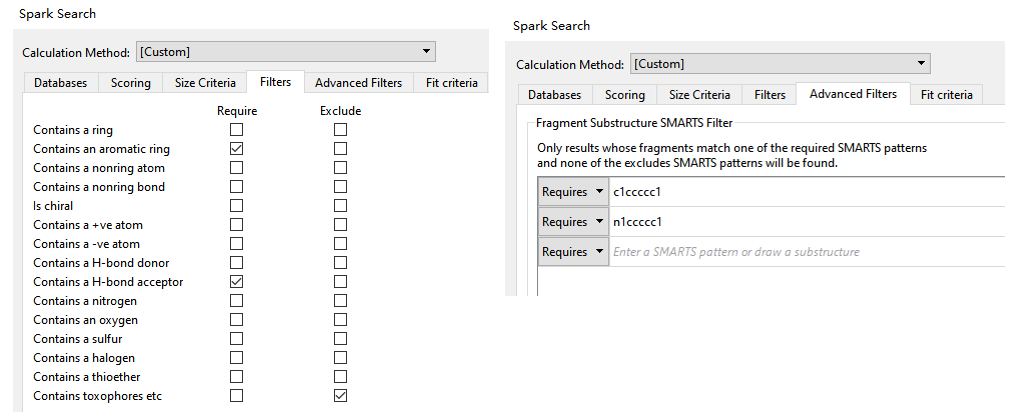
图16. SPARK水分子替换虚拟筛选的过滤器选项
4. 结论
我们用Flare的GIST分析了化合物1c与PD-L1的共晶结构结合位点,结果发现HOH350A是一个高密度、高能、焓为正的水分子,这意味着对该水分子进行替换有可能提高化合物的结合亲和力。鉴于HOH350A与1c的侧链甲氧基距离为6.8Å,进行基团替换时需要考虑在HOH350A与甲基之间填充些形状与静电/药效团互补的取代基,因此进一步用Ligandscout的Apo site grid对HOH350A与甲基之间的子口袋进行药效团特征分析,结果发现芳香相互作用对结合有利。因此确认了1c甲氧基往HOH350A方向生长的假设:1)对HOH350进行替换;2)具有芳香性基团。
有了上述子口袋结合片段的假设之后,用SPARK的水分子替换策略来实现侧链虚拟筛选,并将芳香性基团与氢键相互作用作为约束条件或过滤器来提高准确性。在SPARK生成的500个结果里有43个化合物包含有腈基取代的苯或吡啶,其中包含了文献报道的高活性侧链3-腈基苄基(比如PDB 6R3K配体)与3-腈基吡啶-5-亚甲基(比如PDB 6RPG配体)或它们的衍生物。SPARK非常高效、准确地从数据库中识别出有价值的侧链。
总的来说,结合Flare基于结构设计的策略与Ligandscout对结合位点的药效团特征分析能力,可以为新分子设计提供有力的假设。SPARK的基团替换与骨架跃迁易于将Flare与Ligandscout的假设转化为虚拟筛选查询条件与约束条件,准确地从数据库中发现满足假设的片段、生成具有高价值的新化合物,最终实现高效的分子设计。这为子口袋引入新的相互作用提供了一个高效的通用计算方法。
5. 文献
- Zak, K. M.; Grudnik, P.; Guzik, K.; Zieba, B. J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T. A. Structural Basis for Small Molecule Targeting of the Programmed Death Ligand 1 (PD-L1). Oncotarget 2016, 7 (21), 30323–30335. https://doi.org/10.18632/oncotarget.8730.
- Guzik, K.; Zak, K. M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T. A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60 (13), 5857–5867. https://doi.org/10.1021/acs.jmedchem.7b00293.
- Muszak, D.; Surmiak, E.; Plewka, J.; Magiera-Mularz, K.; Kocik-Krol, J.; Musielak, B.; Sala, D.; Kitel, R.; Stec, M.; Weglarczyk, K.; et al. Terphenyl-Based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein–Protein Interaction. J. Med. Chem. 2021, 64 (15), 11614–11636. https://doi.org/10.1021/acs.jmedchem.1c00957.
- SPARK数据库. http://blog.molcalx.com.cn/2021/04/07/spark-database.html
- Guo, J.; Luo, L.; Wang, Z.; Hu, N.; Wang, W.; Xie, F.; Liang, E.; Yan, X.; Xiao, J.; Li, S. Design, Synthesis, and Biological Evaluation of Linear Aliphatic Amine-Linked Triaryl Derivatives as Potent Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction with Promising Antitumor Effects in Vivo. J. Med. Chem. 2020, 63 (22), 13825–13850. https://doi.org/10.1021/acs.jmedchem.0c01329.
- Flare V6. http://www.cresset-group.com/flare
- Nguyen, C.; Gilson, M. K.; Young, T. Structure and Thermodynamics of Molecular Hydration via Grid Inhomogeneous Solvation Theory. 2011. arXiv:1108.4876v1. https://arxiv.org/abs/1108.4876
- Ligandscout 4.4.8. http://www.inteligand.com
- SPARK V10.6.0. http://www.cresset-group.com/spark


