
用QM构象张力能来对多样的生物等排体母核进行优先级排序
摘要:本文介绍一个基于Flare QM的工作流来对不同母核化合物进行构象张力能计算,包括下面步骤:对化合物进行穷尽构象搜索、在DFT水平对每个构象进行优化、识别出全局极小点并计算全局张力能。本文以Roche报道的四个母核结构多样PDE10A抑制剂为例,从它们与靶标相互作用的角度不能解释实验看到的活性差异,因此构象效应是潜在的原因,用QM计算的构象张力能可对此作出合理解释,并用于指导后续的母核优选工作。我们的工作流重现了文献报道的结果,可以解释实验观察到的活性差异。总地来说,Flare QM可以快速、便利地计算构象张力能,提供坚实的理论支持并用于此类的母核优选场景。
肖高铿/2024-03-20
前言
在基于结构的设计中理解优势、低能构象的重要性是毋庸置疑的1。虽然力场覆盖了药物化学所能访问的大部分化学空间,但是通过对小分子X-ray结构的统计分析可以获得更多、更详细的构象偏好见解2。然而,CSD中覆盖的化学空间与药学上感兴趣的化合物不一定重合,在用CSD分析杂环之间柔性键的扭转角偏好时会出现数据过于稀疏而无法进行统计分析的情况。这时可以选择量子力学进行计算,Roche的Kuhn等人3用PDE10A抑制剂的母核优选案例来对此进行了详细的说明。
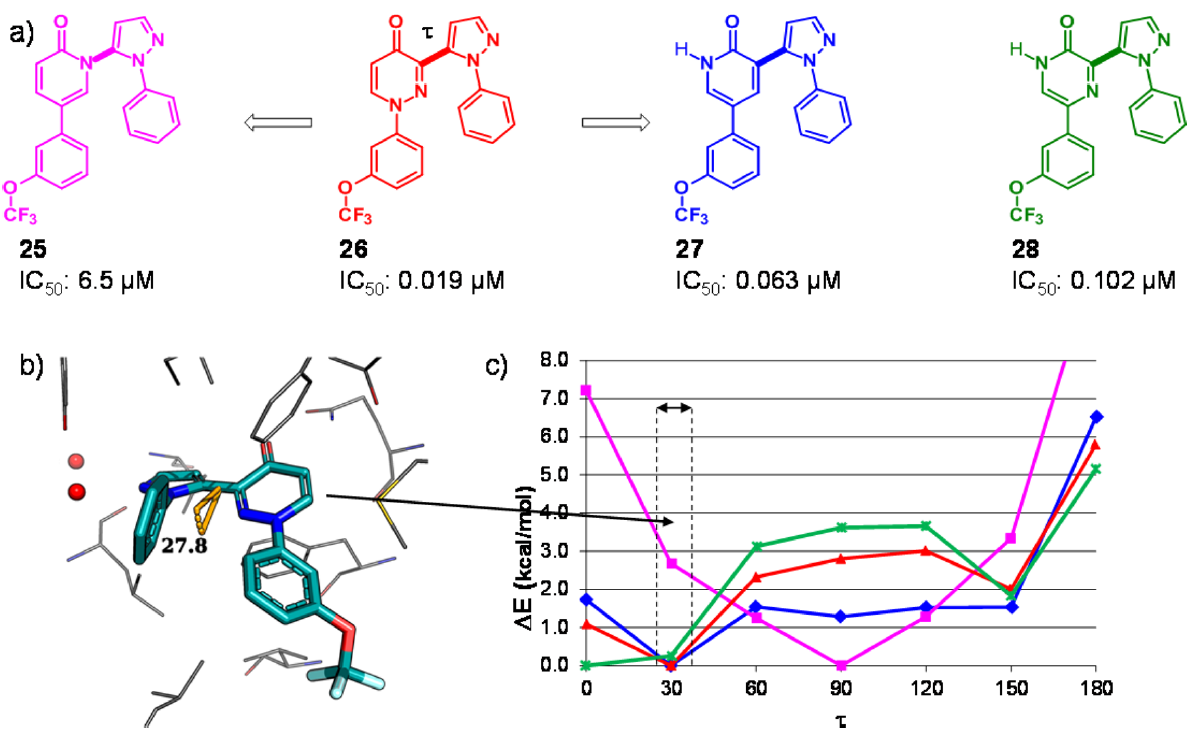
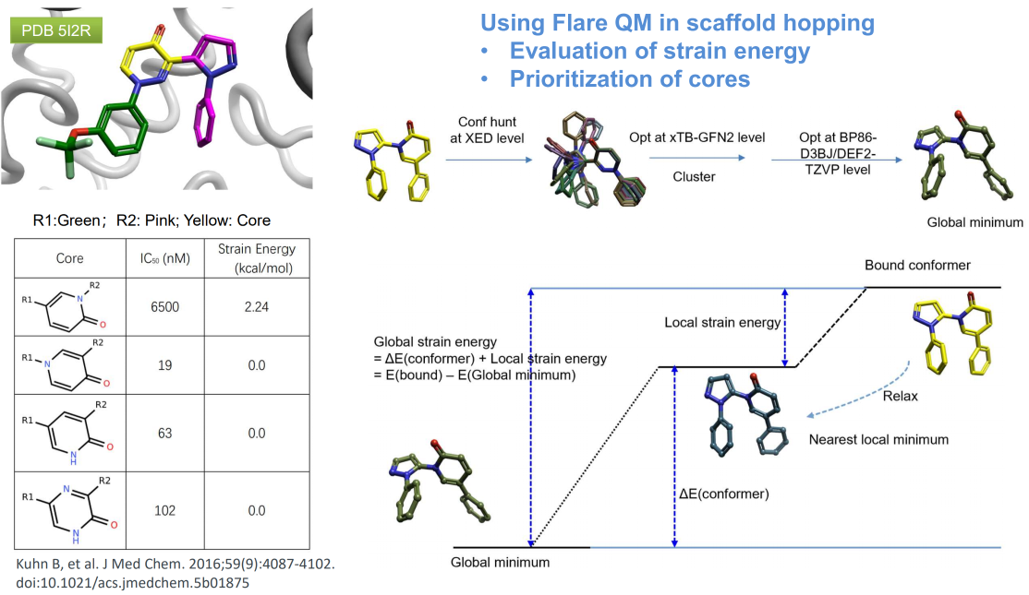
图1.(a)不同母核吡唑类抑制剂对人类PDE10A的IC50值。(b)抑制剂26与人类PDE10A的晶体结构,PDB 5I2R。蛋白质残基和抑制剂分别以灰色和青色显示。(c)用QM方法在B3LYP/cc-pVDZ理论水平对连接两个中心杂芳基的二面角τ进行了柔性扫描。垂直虚线表示在3-吡唑基哒嗪-4-酮母核与PDE10A共晶结构中观察到τ的范围(28-40°)。
在PDE10A项目中,他们解释了3-(吡唑基)哒嗪-4-酮(3-pyrazolylpyridazin-4-one)类抑制剂化合物26(图1)与人类PDE10A的复合物晶体结构PDB 5I2R,揭示了哒嗪-4-酮母核与蛋白的重要相互作用,即羰基氧原子与保守的Gln726侧链的氢键相互作用,以及哒嗪片段与Phe696和Phe729形成的芳香钳之间的π-π相互作用。在26的结合模式中,两个末端苯基形成分子内“边对面”相互作用,吡唑连接臂相对于哒嗪-4-酮平面扭曲约28°(图1)。哒嗪-4-酮母核两个骨架跃迁为两个吡啶-2-酮的变体得到生物等排体化合物25与27(图1),26对PDE10A的抑制作用是27的3倍,是25(图1)的340倍。当与PDE10A结合时,结合模式无法解释25的活性下降,因此怀疑配体的张力能是潜在原因。之后的QM扭转角分析发现两个杂环之间扭转角τ的能量分布存在重大差异。如图1所示,虽然26和27(红色和蓝色曲线)的最小能量与蛋白质结合构象中的扭转角很好地吻合,但25的优势构象却扭曲得多(品红色曲线,在τ≈90°时最小)。25在τ≈30°处有相当大的配体张力能(2-3 kcal/mol),这将转化为结合亲和力下降约100倍。随后合成的化合物证实了这一观察结果。例如,吡嗪-2-酮化合物28的二面角极小值在τ= 0°处,但该化合物仍然有效,这是因为在τ= 30°时,配体张力能仅为0.3 kcal/mol。由于计算的配体张力能与IC50值之间具有良好的定量一致性,因此他们用QM扭转角分析来对不同骨架进行优先级排序,然后再开始合成。
尽管在Roche的PDE10A项目中QM扭转角分析成功的解释了化合物25与26、27以及28的活性差异原因,但是这些化合物的3个芳香环键之间的三个柔性键互相羁绊,严格的扭转角分析需要进行三维扫描,这是一个计算非常昂贵的工作。因此,本文的一个目的是用一个方法简单的构象系综张力能计算方法代替QM扭转角分析,并用来解释25活性降低的原因。另一方面,最近几年报道了一大批性能表现优秀的深度学习打分函数,本文的第二个目的是用此类打分函数预测化合物的结合亲和力,以考察能否优于人工相互作用分析,并尝试与QM扭转角分析组合使用。
材料与方法
结合构象的张力能计算
结合构象的张力能计算如图2所示,张力能为结合构象的电子结构能量减去全局最小点的能量。显然,这个计算方法忽略了熵的贡献,将自由能和能量同等对待。虽然这并不总是对的,但是比较直观且计算简单。

图2.结合构象的张力能计算
为了找到全局最小点构象,先用Flare Conf Hunt & Align对化合物进行构象搜索,对每个构象用QM DFT方法进行能量最小化,用能量最低的那个构象作为参比来计算结合构象的全局张力能。对于结合构象不是极小点的情况,采样冻结两面角的方式进行几何优化,然后进行单点能量计算得到构象张力能。
详细的操作可参见前文用Flare QM计算化合物的ECD图谱,本文不再重述,仅给出参数不同的部分。
蛋白结构与配体结构的准备
将26与PDE10A的共晶结构PDB 5I2R下载到Flare V8,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,并对截短的蛋白质链继进行封端。从准备好的复合物结构里将配体提取出来,得到26的生物活性构象与蛋白结构。在Flare里复制配体26,用原子替换的方式对26进行编辑,并进行能量最小化得到化合物25、27与28。
模型分子的准备
模型分子是从上一步准备好的生物活性构象配体基础上截取得到,截小后的模型分子保留其构象与生物活性构象一致,并作为后续计算的起点。
构象搜索
用Flare 3D-Pose/Conf hunt & Align对化合物25-28进行构象搜索,参数如下:
- Maximum number of conformations: 1000
- Gradient cutoff for conformer minimization: 0.50 kcal/mol/A
- Filter duplicate conformers at RMSD: 0.50 Å
- Energy window: 30.0 kcal/mol
- Acyclic secondary amide handling: Force amides trans
- Remove boats and twist-boats: uncheck
- Turn off Coulombic and attractive vdW forces: uncheck
其中关于酰胺键是否旋转的设置是无效的,因为在计算的分子里,不涉及这样的酰胺键。在默认情况下,Coulombic and attractive vdW forces这一选项是启用的,也就是关闭了长程的静电与vdW吸引作用以加速计算。然而,在我们的这个算例里,在二维结构上长程的苯环之间可能在三维空间是靠近的而具有不可忽略的相互作用,因此需要在构象搜索时考虑这类相互作用。此外,为了得到尽可能多的初始构象,还将能量窗从3kcal/mol调高到30kcal/mol。
QM几何优化
模型分子的QM几何优化使用Flare QM的Minimize conformations工作流,如图3所示,在wB97X-D/DEF2-TZVP//PBE-D3BJ/6-31G+(d)理论水平下计算。
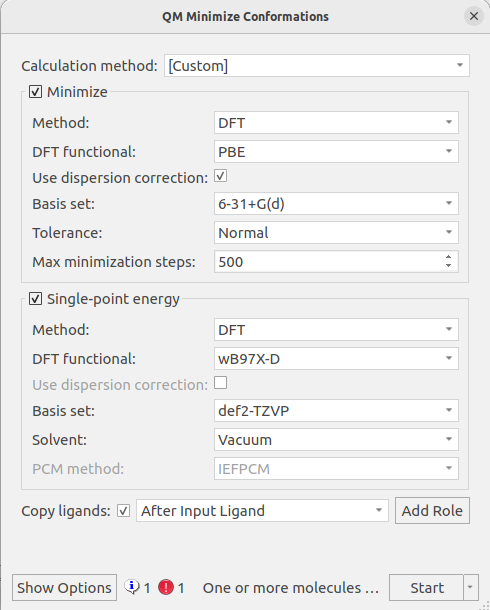
图3. 用Flare QM的Minizize conformations工作流进行几何优化
需要注意的是,这里的优化与能量计算所用的理论模型与Kunh等人3的研究有很大的不同,文献采用的理论模型是B3LYP/cc-pVDZ,而且没有考虑色散效应。
GNINA打分
GNINA是一款采用深度学习CNN模型的分子对接软件6,7,在本文中在score_only模式下用来对化合物25、26、27、28进行打分:
- Affinity(Vina score): Vina打分函数的结合自由能预测值,单位为kcal/mol。
- CNNscore: GNINA的CNN模型pose打分值,表示结合模式正确的概率值。
- CNNaffinity:GNINA的CNN模型结合亲和力预测值,单位为pKd。
对接的输入文件与结果文件见附件。
结果
QM结合构象张力能的计算
我们不直接对化合物25-28进行计算,而是图4简化的模型分子m1、m2、m3、m4来计算。这么做主要是为了在不影响结果的情况下大幅度减少计算量。
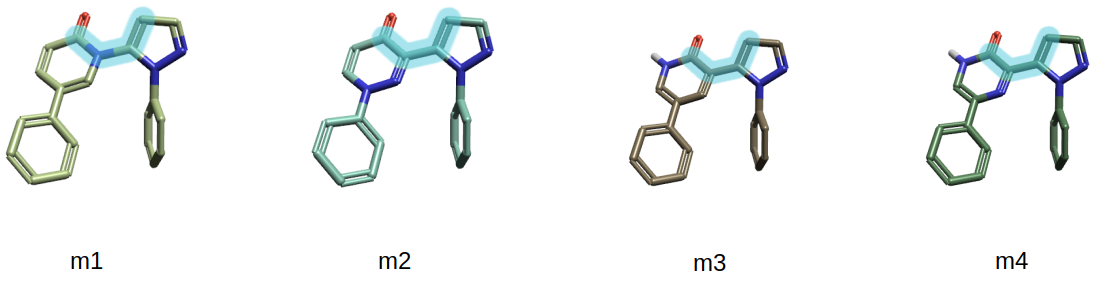
图4. 代替化合物25、26、27、28的模型分子m1、m2、m3以及m4。其中高亮原子定义了Kuhn等人3所述的两面角τ。
模型分子m1、m2、m3、m4用Flare Conf Hunt & Align进行构象搜索,总共分别得到15、15、15、13个构象的构象系综,见表1。接着在PBE-D3BJ/6-31+G(d)理论水平下对构象系综里的每个构象优化,得到的构象进一步在wB97X-D/DEF2-TZVP理论水平下进行单点能计算,得到的电子结构能量用于对构象排序,计算构象张力能。结果如表1所示,4个模型分子优化过后的local minima构象刚好就是全局最小点(global minima)构象,因此它们优化过后的构象张力能(Relaxed Bioactie ΔE)都为0。
表1. 张力能计算结果
| Items | m1/25 | m2/26 | m3/27 | m4/28 |
|---|---|---|---|---|
| Relaxed Bioactive ΔE(kcal/mol) | 0.0 | 0.0 | 0.0 | 0.0 |
| Local Strain(kcal/mol) | 2.24 | 0.0 | 0.0 | 0.0 |
| Global Strain(kcal/mol) | 2.24 | 0.0 | 0.0 | 0.0 |
| Relaxed τ(°) | 53.7 | 32.6 | 33.1 | 26.3 |
| #Conformers | 15 | 15 | 15 | 13 |
| IC50(nM) | 6500 | 19 | 63 | 102 |
在4个化合物中,仅25模型分子m1的生物活性构象不是能量极小点,因此它的张力能计算需要约束三个柔性键的两面角后再进行优化计算(具体方法见附件),然后在wB97X-D/DEF2-TZVP理论水平下进行单点能计算。
其中代表25的m1优化过的两面角τ偏离结合构象最大,生物活性构象为极小点的扭曲,这导致m1的扭转张力能2.24kcal/mol,这与Kunh等人3报道的2-3kcal/mol的张力能是一致的;而28的τ偏离生物活性构象较小,26与27偏离最小,这与它们的IC50排序是一致的。
在Kunh等人3研究中,化合物25全局极小点的τ=90°,而在我们的研究中全局极小点的τ=54°,τ=90°为全局第二最小点。在Kunh等人3的研究中,化合物28全局极小点的τ=0°,而在我们的研究中28全局极小点的τ=26°。化合物25、28全局最小点的构象差异可能与模型分子的结构不同、采用的理论方法不同、优化器、软件实现等有关系,但是计算出来的张力能相差不大。
用分子对接打分函数预测结合自由能
将化合物的IC50转化为结合自由能ΔG之后,如表1所示,可以发现26、27、28的结合自由能相差不到1kcal/mol,这基本是目前计算方法区分活性的极限。化合物25的ΔG明显比其它三个化合物高大约3kcal/mol,这是目前FEP等计算方法可以区分的能力范围之内。用分子对接软件GNINA对4个化合物进行打分,发现CNNscore均大于0.8,这说明这些构象正确的概率比较高,因此进一步考察结合自由预测结果。Vina与CNNaffinity打分函数预测的结合亲和力均不能将活性低的化合物25与活性高的26、27、28区分开来,四个化合物有着一模一样的打分值,这说明CNNaffinity与Vina打分函数可能均未考虑构象效应。
用计算的构象张力能(Strain energy,ΔE)对CNNaffinty进行校正,即:
$$CNNaffinity\_corrected = CNNaffinity + ΔE$$
结果如表1所示,可以发现校正后的CNNaffinity结合自由能预测值与实验值相差很小,最大误差在1.06kcal/mol。张力能校正后的CNNaffinity活性预测能力提高,一方面可能是CNNaffinity本身预测能力不错,只是没有考虑构象张力能;另一方面张力能差异可能是这几个化合物活性差异的单一因素。
表2. 分子对接打分函数预测结合自由能与构象张力能校正
| Items | 25 | 26 | 27 | 28 |
|---|---|---|---|---|
| PDE10A ΔG(kcal/mol)a | -7.07 | -10.53 | -9.82 | -9.53 |
| Vina score(kcal/mol) | -7.91 | -7.91 | -7.91 | -7.91 |
| CNNaffinity(pKd) | 7.39 | 7.39 | -7.39 | -7.39 |
| CNNaffinity(kcal/mol)a | -10.37 | -10.37 | -10.37 | -10.37 |
| CNNscore | 0.83 | 0.83 | 0.83 | 0.83 |
| ΔE(kcal/mol) | 2.24 | 0.0 | 0.0 | 0.0 |
| CNNaffinity-corrected(kcal/mol) | -8.13 | -10.37 | -10.37 | -10.37 |
a:根据亲和力或IC50换算得到。
结论
本文演示了当基于一维扫描的QM扭转角分析方法不适用时的替代方法:对化合物进行穷尽构象搜索、在DFT水平对每个构象进行优化、识别出全局极小点并计算全局张力能。以Roche报道的四个含有多个柔性键的分子为例,用Flare QM重现了文献报道的计算结果,可以解释实验观察到的活性差异。无论是传统的Vina打分、还是深度学习CNN打分,均不能将结合自由能相差3kcal/mol的两组化合物的区分开来,但是用张力能校正后的CNN打分可以。总地来说,当相互作用不能解释活性差异时,构象效应是潜在的原因,Flare QM可以快速、便利地计算构象张力能,为母核优选提供坚实的理论支持。
附件
1. GNINA对接打分计算与输入文件:docking.tar.gz,内容包括:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 | docking ├── 25.log ├── 25_out.sdf ├── 25.sdf ├── 26.log ├── 26_out.sdf ├── 26.sdf ├── 27.log ├── 27_out.sdf ├── 27.sdf ├── 28.log ├── 28_out.sdf ├── 28.sdf ├── 5i2r_prot.pdb ├── 5i2r_prot.pdbqt └── dock.conf 0 directories, 15 files |
2. 25的模型分子m1约束两面角的优化计算文件:m1_bioactive_conformer_opt.dat。
参考文献
- Groom, C. R.; Allen, F. H. The Cambridge Structural Database in Retrospect and Prospect. Angew. Chem. Int. Ed. 2014, 53(3),662–671.
- Brameld, K. A.; Kuhn, B.; Reuter, D. C.; Stahl, M. Small Molecule Conformational Preferences Derived from Crystal Structure Data. A Medicinal Chemistry Focused Analysis.
J. Chem. Inf. Model. 2008, 48 (1), 1–24. - Kuhn B, Guba W, Hert J, et al. A Real-World Perspective on Molecular Design. J Med Chem. 2016;59(9):4087-4102. doi:10.1021/acs.jmedchem.5b01875
- Flare V8. https://www.cresset-group.com/software/flare
- Trott, O. and Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 2009, 31(2), 455–461. https://doi.org/10.1002/jcc.21334.
- Sunseri, J., & Koes, D. R. Virtual Screening with Gnina 1.0. Molecules, 2021, 26(23), 7369. https://doi.org/10.3390/molecules26237369
- GNINA 1.0, https://github.com/gnina


