
摘要:罗氏的科学家们分享了一系列小分子药物发现案例,在这些案例中计算方法前瞻性地影响了罗氏的研究项目,目的是凸显那些能够实际带来附加值的方法。本文简述了应用于各种酶和受体的方法和技术,其中大多数是关于对分子构象和相互作用知识恰如其分的应用:填充亲脂性口袋以获得亲和力或选择性、添加极性取代基、骨架跃迁、SAR转移、构象分析和分子叠合。其中一个案例是序列驱动的集中筛选,阐明了适当的信息预处理是如何使高效地利用先验知识成为可能。他们的结论是,使化学家能够专注于化学空间中有希望的区域的定性陈述往往比定量预测更有效。
编译:肖高铿 2020-01-25
原文:Kuhn B, Guba W, Hert J, et al. A Real-World Perspective on Molecular Design. J Med Chem. 2016;59(9):4087-4102. doi:10.1021/acs.jmedchem.5b01875
前言
在当前的小分子药物发现中,分子设计的作用似乎已经牢固确立。 有关药物化学的期刊文章通常会参考使用模拟的结合模式,虚拟筛选或预测的分子性质。全球的制药公司都报告了其部署和使用工具的方式1-3。
然而,分子设计对单个项目进展的真正贡献很少在公开场合进行辩论和剖析,令人信服的案例研究比分子模拟软件所暗示的大量贡献要稀缺得多。 通过罗氏(Roche)十多年来药物发现的一系列项目简介,我们说明了哪些工具和方法在过去几年中取得了最丰硕的成果。我们证明了这些方法背后有共同的原则,这使我们认为,药物发现中分子设计的首要目标可以以一种让人更自然地关注有效活动的方式重新定义。
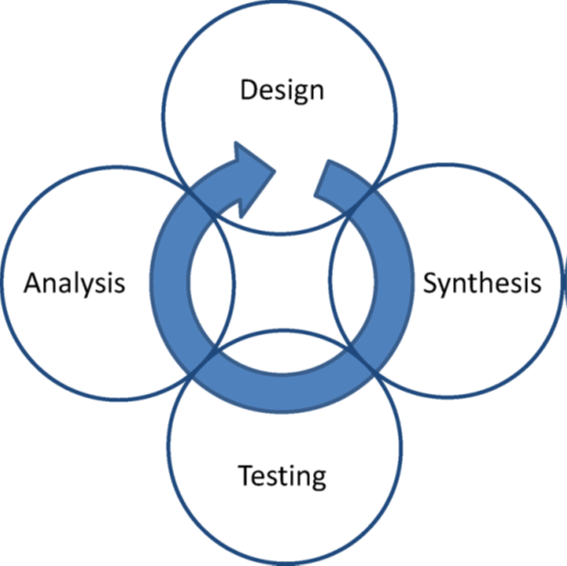
Figure 1. 经典的设计周期示意图。合成和测试为实验组件,分析和设计为概念组件。设计区域中的箭头是断开的以说明从已知到未知以及虚拟是离散步骤。本文涉及设计的各个方面,这些方面可以采用完全不同的形式,具体取决于新思想(idea)产生和过滤方式。
术语“分子设计”与广泛接受的设计周期概念密切相关,这意味着药物发现是定向进化的过程(图1)。该循环可细分为合成和测试的两个实验部分,以及一个概念阶段。这一概念性阶段开始于数据分析,结束于合成下一轮化合物的决策。在分析和决策之间会发生什么是不太明确的,我们将其称为设计阶段。在任何实际项目中,设计阶段都是一个多方面的过程,结合了有关项目状态和目标、先验知识、个人经验、创意和关键过滤要素以及实际规划等信息。据我们了解,分子设计的任务是将这一复杂的过程尽可能地转变成一个明确的、合理的和可追溯的过程。任何分子设计方法实用性的两个关键标准是:产生实验可测试的预测,以及无论这些预测最终正确与否,实验结果应该增加对可用优化空间的理解,因此以迭代的方式提高正确预测的机会。分子设计的主要成果是一个想法(idea)4,成功是对研究生物系统的改良化合物的有意义的贡献。
Table 1. 本文的案例概述
| Project | Approach | Impact |
|---|---|---|
| DPP-IV | Filling a lipophilic pocket | Affinity increase while balancing polarity |
| FABP 4/5 | Targeting specific residue differences in the binding site | Affinity and selectivity increase |
| Cathepsin S/L | Assessment of electrostatic complementarity of protein and ligand. Visualization of halogen bonding potential | Affinity and selectivity increase |
| Β-Tryptase | Scaffold hopping | Alternative scaffold |
| BACE1 | Scaffold hopping | Solubility increase, alternative scaffold |
| PDE 10A | Targeting binding site hotspots identified from multiple crystal structures, Calculation of torsion energy potentials | Affinity increase,Prioritization of alternative scaffolds |
| HSL | Hypothesis derived from homology model | Novel scaffolds with potential for improved PK properties |
| GPBAR1 | Hypothesis derived from homology model | Targeted introduction of solubilizing group |
| L-CPT1 | Bioactive shape hypothesis | Prioritization of scaffolds |
| SSTSR | Sequence-based binding site similarity as a basis for focused screening (“chemogenomics”) | Identification of druglike hits |
下面,我们报告10个案例研究用来说明分子设计在罗氏药物发现项目中的贡献。表1为这些项目的概述,每一个都以简短的项目基本原理、问题陈述开始,然后是所采用的计算方法的描述和所获得的结果的讨论。所有研究均以前瞻性方式进行;在准备计算输入和规划药物化学时,不知道实验结果。大部分研究以前都没有发表过。因此,本研究展示了一组新化合物以及与之相关的13项PDB数据。在阅读本案例时,应该记住它们不是完整的项目描述,而是专注于一个学科的作用。这意味着有时引入新化合物而不描述它们的起源,以便将注意力集中在一个特定阶段和设计问题上。这也意味着并非优化的所有方面都受到相同程度的关注。例如,ADMET性质的优化在本文所涉及的所有项目中都发挥了突出的作用,但在案例研究中并非总是如此。
案例
分子设计的起源与更常规的晶体结构测定的出现是一致的,并且本质上与该信息的利用同义,一次一个结构。 如今,通过将此类分析扩展到数千个小分子和蛋白质复合物结构上,可以对蛋白质和小分子的相互作用和构象偏好做出可靠的经验性陈述。我们和其他人已经汇编了此类知识5-7,在此我们将不再尝试进行新的总结。 相反,我们想思考一下如何将这些知识用于设计目的。
设计配体以优化其与亲脂性口袋的形状互补性以获得亲和力或选择性。 “口袋填充”可能是最显而易见、应用最广泛的设计概念,但正如以下三个应用案例所示,这并不意味着其中没有挑战。
DPP-IV抑制剂
二肽基肽酶IV(DPP-IV)抑制剂已成为治疗2型糖尿病患者的推荐疗法8,9。内部高通量筛选(HTS)发现的2-aminobenzo[a]quinolizine类苗头化合物1是一种DPP-IV抑制剂,仅中等活性,但具有令人感兴趣的类药特性(图2)。在DPP-IV晶体结构公开之前,通过随机化学修饰改善结合亲和力的尝试都不成功。进入项目大约1年后,化合物1与人类DPP-IV的结合模式通过共晶结构得以揭示,理性设计的方法才得以进行(图2a)。
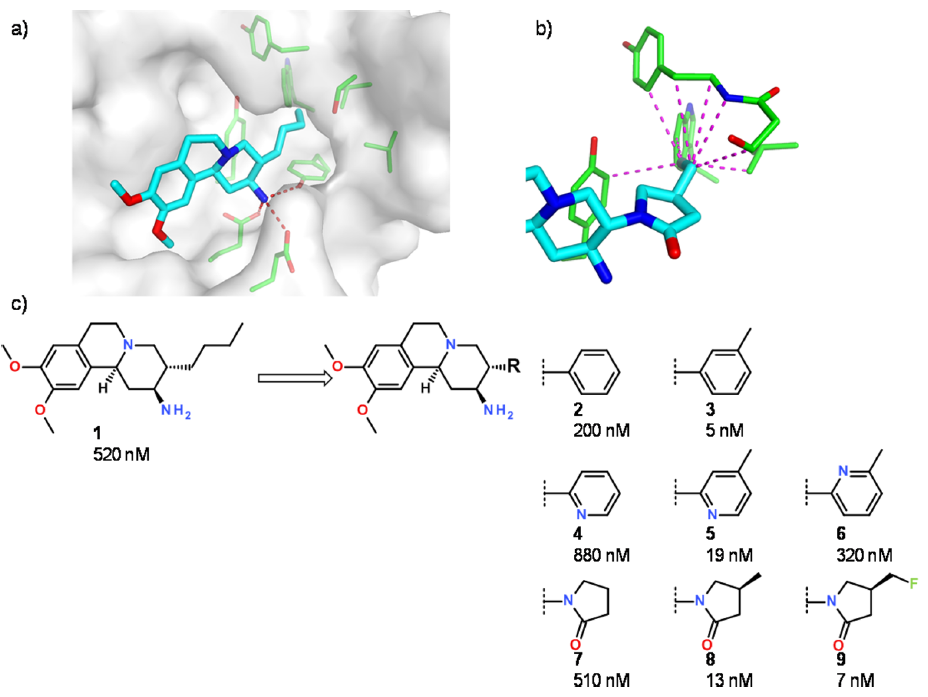
Figure 2.(a)人DPP-IV与抑制剂1的晶体结构(PDB 3oc0)。形成S1口袋的蛋白残基、与化合物1胺基发生氢键相互作用(红色虚线)的侧链以绿色突出显示,抑制剂以青色显示。(b)人DPP-IV与化合物9的晶体结构(PDB 3kwf)S1口袋的特写图。品红色的线表示氟甲基与蛋白质之间的距离小于等于4.0Å的非键接触。(c)所选化合物对DPP-IV的SAR11,14,数值是化合物对人DPP-IV酶抑制的IC50值。
结合位点的分析表明,相当柔性的正丁基指向DPP-IV由几个芳香族和脂肪族氨基酸的侧链组成的S1口袋。 S1口袋的高度亲脂性以及脯氨酸环系被DPP-IV底物所识别这一事实表明:用更刚性的环系替换正丁基可以显着提高亲和力。用苯基(化合物2)或吡啶(化合物4)进行了标准药物化学替换,虚拟筛选和分子设计技术补充了诸如N-取代的内酰胺7。令人惊讶的是,这些替代并没有显着改善DPP-IV的结合亲和力。通过详细分析环状衍生物与S1口袋的形状互补性以及与另一种DPP-IV抑制剂系列(其P1取代基进行了优化)的叠合模拟10,发现芳族间位的有额外空间可用。如化合物3、5、8和9所示,通过小的甲基和氟甲基取代,可以实现很高的结合亲和力提高(40至70倍)。吡啶类似物6的DPP-IV活性大大降低:氮孤对电子与酪氨酸残基的π电子云之间不利的相互作用可能抵消了引入甲基所带来的亲和力增益。对DPP-IV底物衍生物的模拟表明:极性官能团仅是邻位容许,该位置的极性基团暴露于溶剂和极性蛋白质残基。虽然化合物3、5、8和9具有相似的DPP-IV亲和力,但8和9由于两亲性的降低而显示出最佳的类药性,这是由于极性基团的分布更加平衡所致11。最后,选择了化合物9(Carmegliptin)进入临床开发。
DPP-IV的例子说明,当有晶体结构的结合模式可用并且界定清楚的疏水性口袋很少被占据时,可以快速实现活性增强。在这种情况下,单个重原子就可获得惊人的亲和力增加,从而证实了在包埋腔内填充小空隙的重要性12,13。DPP-IV S1口袋的亲脂性高且柔性低,这使其成为基于结构设计的理想案例。当然,单独的高亲和力并不是优化的目标;在本案例中,极性的定向引入对于平衡PK特性和消除hERG抑制至关重要。次优的形状互补性可以容易地检测到并可视化;因此,对3D复合物结构进行适当的注释和可视化使得基于结构的设计可以用简单的方法来实现。由于(去)溶剂化能的不确定性以及蛋白质的诱导契合,加上大部分极性相互作用的高度定向性,使得在更具极性或更柔性的结合口袋中优化抑制剂的亲和力更具挑战性。
Fatty Acid Binding Protein 4/5(FABP 4/5)
脂肪酸结合蛋白(FABPs)是具有组织特异性分布的胞质脂质结合蛋白,与脂肪酸的摄取、代谢和细胞内运输有关15。根据流行病学研究和动物基因敲除模型,同工型FABP4和FABP5已被确定为潜在的糖尿病和动脉粥样硬化靶标16。理想的抑制剂特性包括对FABP3具有高度选择性。对Roche化合物库一组1200个化合物的集中库筛选得到喹啉10,对FABP4(0.1μM)具有良好的活性,但对FABP3没有选择性、对FABP5没有检测到活性。因此,我们的目标是改善对FABP4和FABP5的活性,同时强烈降低对FABP3的活性。

Figure 3.(a)化合物10和13与FABP4的结合模式(绿色,PDB 5edb和5edc)。 左图显示了FABP3、FABP4和FABP5结合位点里具有差异的氨基酸。 FABP3(青色)具有Leu104、Leu115、Leu117(PDB 2hmb),而FABP4(绿色)具有Ile105、Val116、Cys118(PDB 5edb),FABP5(品红色)具有Ile107、Val118、Cys120(PDB 1b56)。 5edb中的甲基和5edc中的哌啶基的电子密度相当弱。Cys118侧链和配体哌啶基的两个构象用于模拟FABP4-13复合物结构的电子密度。(b)化合物10-13对FABP3、FABP4和FABP5的抑制常数(Ki)。
解决此问题的常用方法是将相关靶标上的序列信息映射到配体结合位点上,并靶向3D空间中显示氨基酸差异的那些区域。在项目早期就解释了FABP4与喹啉苗头化合物10的复合物晶体结构,发现有一个区域,其中FABP3与其他两个同工型之间存在三个残基差异的簇(图3a)。由于FABP3在此簇为三个Leu侧链,这比FABP4和FABP5中的Ile、Val和Cys残基更大,因此对于FABP3来说该口袋似乎更小。因此,增加喹啉2-位取代基的尺寸(甲基、乙基、异丙基、哌啶基的尺寸依次增大)会引起与FABP3的空间碰撞。如图3所示,此策略导致FABP3抑制常数从90nM急剧增加至10μM,与2位取代基的大小增加密切相关。由于FABP4和FABP5的结合口袋没有被原始甲基最佳填充,因此较大的取代基进一步改善了这两种同工型的配体亲和力。对于FABP5效果尤为明显,经过引入哌啶基,苗头化合物10由检测不到的活性转变为具有亚微摩尔抑制活性的化合物13。在13与FABP4形成复合物的晶体结构中(图3a),喹啉哌啶以假轴构象嵌入选择性口袋中。先前基于CSD的构象分析表明5,这对于与缺电子芳环(比如吡啶或嘧啶)连接的哌啶并不罕见,这表明这种情况下的配体张力能很小。Cys118周围的电子密度必须用两个旋转异构体来描述,而相邻配体哌啶基的两个假轴构象是这两个旋转异构体所必需的。
靶向结合位点中的氨基酸差异通常可以通过聚焦直接的蛋白质-配体接触来实现,例如通过设计与靶标原子发生有利相互作用而与反靶标发生立体碰撞来实现降低对反靶的结合能力。尝试”突破界限”也是很好的实践,也就是设计与蛋白质口袋碰撞的分子以测试其柔性。以这种方式发现新的蛋白构象17,18。
不幸的是,这种情况通常不如在FABP3情况下有利。在FABP3案例中,引入与不希望的靶标的简单立体碰撞就足以实现同工型的特异性。更精细的构象可塑性和间接作用可能是驱动靶标选择性的主要因素,而这些实际上几乎不可能被晶体学检测到或被计算预测出来。在其他情况下,无法通过利用与配体紧邻的氨基酸差异来实现选择性。 如下一个示例所示,将离结合位点更远的区域考虑进来并利用分类的相互作用等替代方式会有所帮助。
Cathepsin S/L
半胱氨酸蛋白酶组织蛋白酶S(CatS)和L(CatL)双重抑制是靶向肾脏肾小球疾病的潜在策略。 在先前项目中发现了有效的、选择性的CatS抑制剂后19,该团队着手开展了一项重点项目,旨在调整其对CatL活性。 项目的起点是四氢吡咯化合物14(图4),其腈基与活性位点的半胱氨酸共价结合,而取代的苯砜和氯取代的吡啶分别占据S2和S3口袋。由于CatS的体外活性已经处于低纳摩尔范围内,因此设计目标是特异性改善CatL结合,同时保持对其他组织蛋白酶的良好选择性。
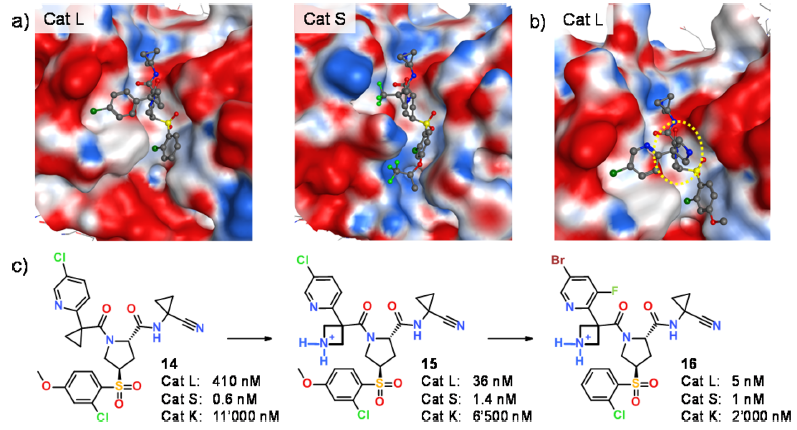
Figure 4. (a)人组织蛋白酶L(左,PDB 2yjc)和小鼠组织蛋白酶S(中,PDB 4bpv)结合位点的表面静电势。出于技术原因,将小鼠晶体结构作为人类同工酶的替代物。在化合物14 7Å范围内带电荷的氨基酸有:人CatL(2 Glu,4 Asp),人CatS(1 Asp,1 Lys),小鼠CatS(1 Glu,1 Asp,1 Lys)。 表面静电势用MOE计算20,色标范围从-40 kcal/mol(红色)至+40 kcal/mol(蓝色)。(b)化合物15与人类CatL的X射线共晶体结构(PDB 5f02)。新引入的氮杂环丁烷环以黄色突出显示。(c)对人组织蛋白酶L,S和K的IC50值19
在CatS / L示例中,在更全局的水平上分析结合位点的差异是很有帮助的。如图4所示,组织蛋白酶L和S的静电势存在显着差异20。CatL结合位点主要为负的表面静电势:在化合物14的约7Å半径范围内,有六个酸性Asp和Glu残基,但在人的CatL中没有碱性的Lys或Arg残基。相反,CatS的结合位点更加均衡。人类的CatS位点只有一个Asp残基与一个Lys残基互补。在此分析中,相关的半胱氨酸蛋白酶组织蛋白酶K(CatK)介于CatL和CatS之间,其人类酶的结合位点有3个Asp而没有Lys/Arg残基。
为了优化与组织蛋白酶L的静电互补性,中性环丙基接臂被带正电的氮杂环丁烷取代。由于静电相互作用的长程性质,我们推断引入远离(大于6Å)带负电荷的蛋白质侧链的正电荷配体对CatL结合可能是有利的。如图4中的一对化合物14、15所示,该化学变化确实导致对CatL的结合亲和力增加11倍,而对CatS和CatK的IC50值仅改变了2倍。随后CatL-15的复合物晶体结构表明,氮杂环丁烷环保持溶剂暴露状态,并且仅通过水介导的接触与蛋白负电荷相互作用(图4b)。在先前对CatL S3口袋中的卤键进行了系统研究的基础上21,我们将化合物15的5-氯吡啶用5-溴-3-氟吡啶替换得到化合物16以进一步选择性地优化对CatL的活性。总之,通过优化静电互补性和微调卤键相互作用,识别出几乎等价的CatS/L双重抑制剂,对其他组织蛋白酶具有良好的选择性。
本研究举例说明了:不仅与蛋白原子直接接触的配体基团可以用来优化结合,而且溶剂暴露的配体基团也可以用来优化结合22,23。改造配体及其蛋白质环境的静电互补性是获得亲和力并提高对反靶选择性的有用策略24,25。静电性分析应是研究新蛋白质结合位点的常规步骤。
迄今为止,我们一直关注优化相互作用以获得亲和力和选择性。在基于结构的设计工具的另一端,有一些方法旨在修饰分子母核的同时而保持现有的相互作用。这样的技术被称为骨架跃迁(scaffold hopping)方法。 ReCore26,27是建立在CAVEAT28原始概念上的一种这样的方法。ReCore通过识别小分子晶体结构中与出口载体(exit vector)几何匹配并可选地满足药效团约束的片段来进行骨架替换。 接下来的两个示例说明:骨架跃迁可以成为访问全新化学空间的强大工具。
β-Tryptase
β-胰蛋白酶是一种肝素稳定化的四聚体丝氨酸蛋白酶,主要在肥大细胞中表达,在呼吸道炎症和过敏反应中起到关键作用。赛诺菲-安万特公司的参比抑制剂在我们的发现项目启动时就作为强效胰蛋白酶抑制剂,该抑制剂在S1口袋中有个苄胺[29],一个哌啶酰胺骨架连接臂,以及在S4口袋中有各种取代的芳环(图5)。 设计的目标是发现拥知识产权、强效和选择性的β-胰蛋白酶配体。
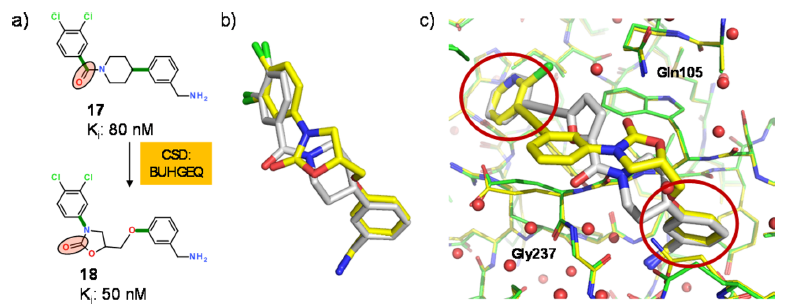
Figure 5. (a)胰蛋白酶参比抑制剂和ReCore26得到的新型恶唑烷酮骨架。 用于定义链接区的键加粗显示为绿色的棒,ReCore约束的受体官能团以红色椭圆突出显示。(b)ReCore在骨架跃迁过程中产生的叠合。(c)参比化合物-人β-胰蛋白酶共晶(白色,PDB 4a6l)和新设计的恶唑烷酮抑制剂-人β-胰蛋白酶共晶(黄色,PDB 5f03)叠合。末端基团用红色圆圈突出显示,以强调整体结合模式的相似性。
赛诺菲-安万特(Sanofi-Aventis)分子之一与人 β-胰蛋白酶的复合物晶体结构在项目早期就已经解释出来。如胰蛋白酶样丝氨酸蛋白酶所预期的那样,碱性苄胺基占据了S1口袋与阴离子Asp207侧链相互作用,而取代的芳环指向诱导型S4口袋。连接这两个部分的配体片段具有相对小的蛋白质接触。ReCore用来识别连接P1和P4芳基的新连接基团,同时保留哌啶酰胺的受体官能团(图5a)。将200个打分最高的ReCore结果聚类得到结构多样的子集,并且在胰蛋白酶结合位点进行可视化分析。在对一些提出的骨架进行修饰以使其具有更好的结构匹配性和分子性质后,与药物化学家讨论了20种骨架的选择。由于其对 β-胰蛋白酶的新颖性以及相对容易合成,CSD骨架BUHGEQ被优先选择。新设计的噁唑烷酮分子对人 β-胰蛋白酶显示出两位数nM水平抑制活性,并对相关蛋白酶具有普遍的高选择性。虽然成功的骨架替换表明我们的叠合假设是正确的,但 β-胰蛋白酶-噁唑烷酮衍生物的共晶体结构显示出中心片段的惊人翻转(图5c)。新连接臂的构象如所预测的一样,但反转为镜像。这是由于形成了可供选择的氢键:哌啶酰胺羰基与Gly237主链氮之间较弱的相互作用被与Gln105的侧链酰胺的氢键相互作用取代。这一发现提醒我们永远不要相信结合模式假设,除非已经探索了所有可能的替代方案!
BACE1
我们之前已经描述了多种环状脒类BACE1抑制剂30,31,这是一个广为研究的头部基团32。脒片段部分通过三个氢键与催化位点的天冬氨酸残基Asp93和Asp289紧密结合,并提供了合适的出口载体以延伸至S2’以及S1和S3子结合位点。 S1的口袋很浅,我们的抑制剂含有一个间位取代苯环的P1取代基,它将头部基团与P3取代基连接起来30。
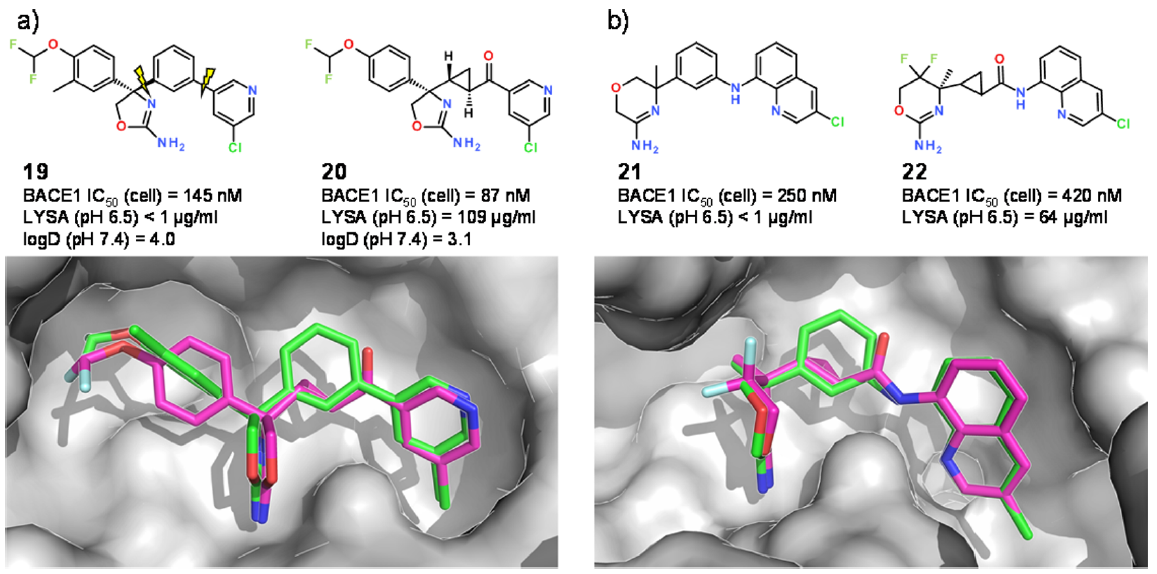
Figure 6.(a)19中的P1苯环被20中的反式环丙基酮所取代。19(PDB 5ezz)和21(PDB 5ezx)的晶体结构的叠合表明P1苯环与其模仿设计之间完美叠合。(b)反式环丙基酰胺是间位取代苯胺的模仿设计:化合物21(PDB 5f00)和22(PDB 5f01)的叠合结果。
我们正在寻找取代P1苯环的替代品以改善物理化学性质。 图6中的化合物19高度亲脂性,log D为4.0,因此动力学溶解度非常低。ReCore建议使用反式环丙基酮作为间位取代苯环的极性替代品(CSD entry:FUQGAZ)。出口载体的二面角仅相差2.6°,几乎完美地叠合。将该片段插入到先前的化合物中得到化合物20。20在细胞水平的活性测试中仅略微活性增加,但其降低了的log D和增加了的溶解度显著地改善了理化性质(图6a)。
芳香胺因其可能与反应性代谢物的形成以及遗传毒性有关而在药物化学中备受关注33。为了探索使用反式环丙基酮替代间位取代苯胺的适用性,我们将化合物21的苯胺替换后生成22。与之前的情况一样,我们发现建模的和实验确定的受体结合构象之间完美匹配,同时改善了溶解度(图6b)。
β-胰蛋白酶和BACE1实例表明,使用CSD-衍生的连接臂进行骨架替换可以识别非显而易见的替代物,从而为项目打开了新的化学空间。适合使用该方法的结合位点在分子的远端具有主要配体识别模式,而在中心区域几乎没有直接的蛋白接触。这允许实现大量不同的设计方案并具有更大的设计限制容差。在不满足理想情况的条件下,约束条件的数量可以是一个限制条件,在非常窄的搜索空间内,骨架替换搜索本质上就是药效团搜索。在这种情况下,将已知抑制剂的片段互相组合与连接的方法可能更有前途。不同共晶结构的3D片段拼合是一种组合不同系列分子感兴趣片段的巧妙方法。结构信息越大越多样化,产生新颖组合的机会就越大。BREED算法是最早使用项目结构进行3D拼合34的方法。将片段筛选的苗头化合物与其它激酶抑制剂的已知片段组合设计全新的DDR1/2激酶抑制剂是该技术巧妙应用的例子35。在下面的例子中,卤素原子从一个系列转移到另一个系列,但是该概念同样适用于更大的配体片段。
PDE10A
因其在纹状体中的患病率很高,并且临床前数据显示出抗精神病药和预认知作用,人们认为抑制磷酸二酯酶10A(PDE10A)是治疗精神分裂症有吸引力的靶点36。我们鉴定了几种三唑并嘧啶与三唑并吡啶化合物是强效、选择性的PDE10A抑制剂(Figure 7)。在本研究中我们阐明了如何通过迁移不同系列的知识来改善结合亲和力。
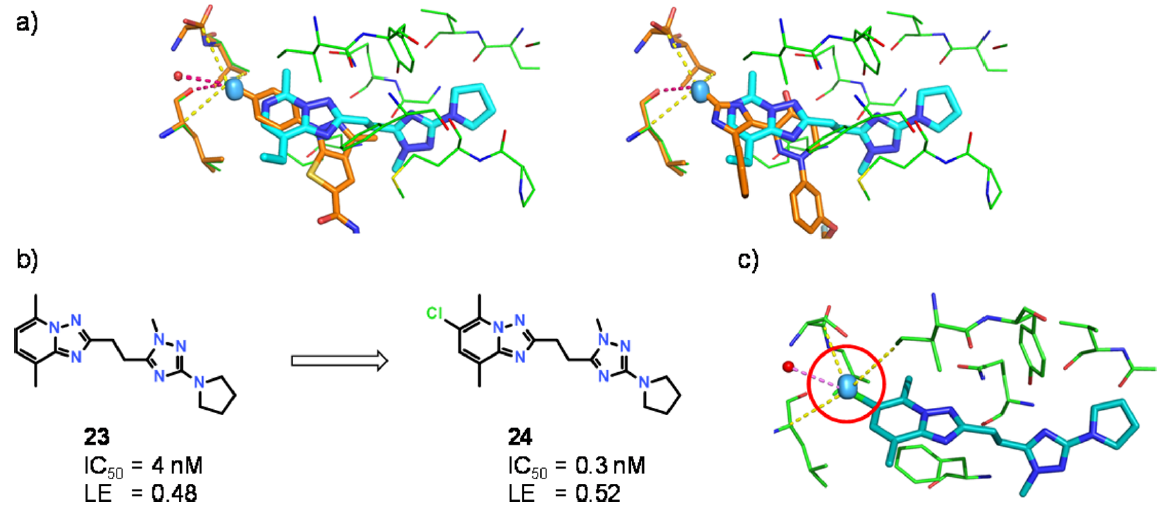
Figure 7.(a)人类PDE10A(绿色)与三唑并嘧啶抑制剂(青色,PDB 5edh)的的晶体结构。橙色是Roche PDE10项目其他两个抑制剂的叠合结构,根据Scorpion网络分析39(PDB 5ede和5edg),它们的氯原子占据一个结合热点。热点显示为青绿色椭圆形,检测到的有利相互作用用虚线表示(黄色:色散;粉色:卤键)。(b)三唑并吡啶23和24对人类PDE10A的抑制常数与配体效率(LE)。(c)化合物24与人类PDE10A的晶体结构(PDB 5edi)。结合热点用红色圆圈突出显示。
配体模式(Motif)从一个系列转移到另一个系列的前提条件是:有晶体结构可以进行叠合,并且可以通过分子间相互作用的分析或通过活性悬崖映射到结合位点来理解结合位点的“热点” 37。基于Proasis38中实现的自动叠合流程和Scorpion对有利相互作用的网络分析39,我们预测了PDE10A活性位点的热点。Scorpion打分贡献大于1.5的配体原子被标记为热点,并根据其空间分布进行聚类。然后将每一类(Cluster)可视化为椭圆形,其形状代表着该类成员的分布。我们识别出了一个具有良好色散与卤键等有利相互作用的卤原子结合热点,如其他抑制剂系列化合物中的两个与PDE10A复合物晶体结构所示的那样(图7a)。三氮唑并嘧啶与PDE10A复合物晶体结构的叠合表明,三氮唑并吡啶系列通过6-位取代可以到达此热点。化合物23的Cl取代衍生物不仅对PDE10A的抑制作用提高了10倍以上,而且配体效率也提高了。化合物24与PDE10A催化结构域的复合物晶体结构证实:氯原子的加入确实占据了聚类的椭球体并产生了预期的相互作用(图7c)。尽管这种情况很特殊,将Cl取代基从一个系列迁移到另一个系列的看起来很简单、直接,但此处描述的工作流程却更为通用,可以从许多晶体结构中吸取教训,并迁移更大的配体片段。
毫无疑问,在基于结构的设计中理解优势、低能构象的重要性是毋庸置疑的40。虽然当今的力场覆盖了药物化学所能访问的大部分化学空间,但通过对小分子X-ray结构的统计分析可以获得更多、更详细的构象偏好见解5。但是,CSD中涵盖的化学空间不一定与药学上感兴趣的化合物重合。一个具体的例子是分析在CSD中代表性不足的杂环之间的扭转角。 当实验数据过于稀疏而无法进行统计分析的时候,可以选择量子力学进行计算。
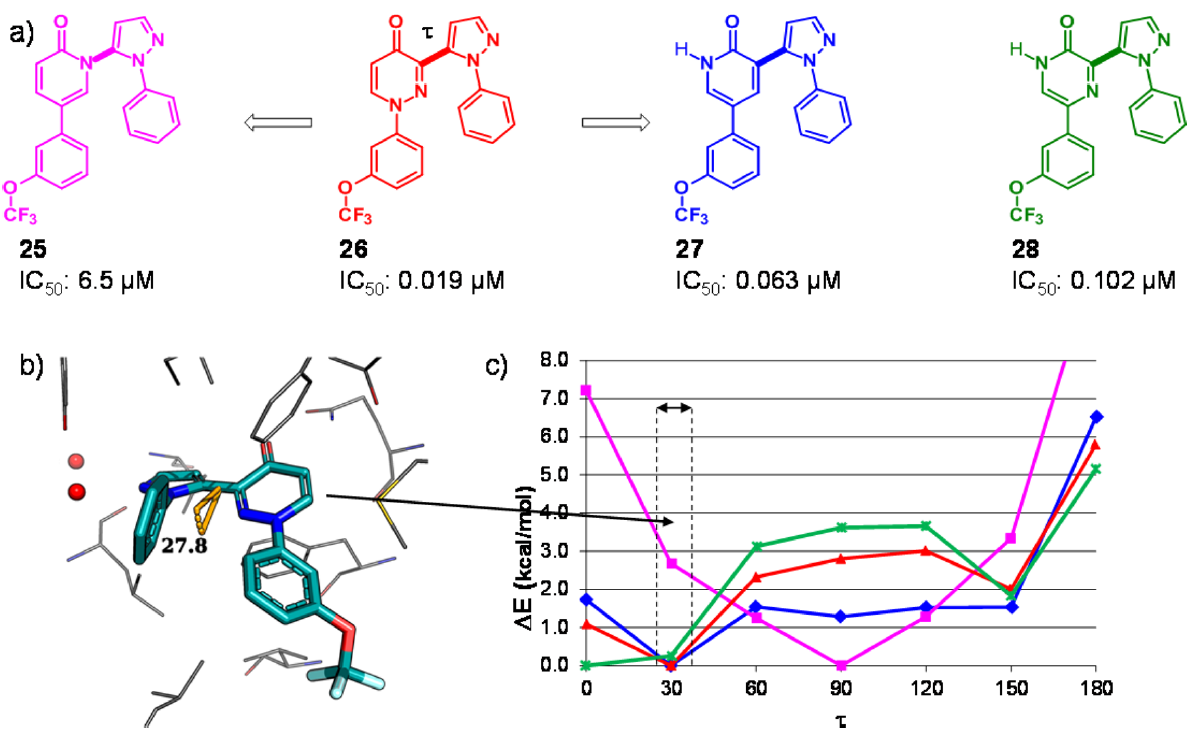
Figure 8.(a)具有吡啶-2-酮、哒嗪-4-酮或吡嗪-2-酮母核的吡唑抑制剂对人类PDE10A的IC50值。(b)抑制剂26与人类PDE10A(PDB 5i2r)的晶体结构。蛋白质残基和抑制剂分别以灰色和青色显示。(c)对连接两个中心杂芳基的二面角τ进行了柔性扫描,用Gaussian 9841在B3LYP/cc-pVDZ理论水平进行计算。垂直虚线表示在3-吡唑基哒嗪-4-酮母核与PDE10A共晶结构中观察到τ的范围(28-40°)。
作为PDE10A项目的一部分,我们研究了HTS发现的3-(吡唑基)哒嗪-4-酮(3-pyrazolylpyridazin-4-one)类抑制剂(26)。化合物26与人类PDE10A的复合物晶体结构揭示了哒嗪-4-酮核心与蛋白的重要相互作用,即羰基氧原子与保守的Gln726侧链的氢键相互作用,以及哒嗪片段与Phe696和Phe729形成的芳香钳之间的π-π相互作用。在26的结合模式中,两个末端苯基形成分子内“边对面”相互作用,吡唑连接臂相对于哒嗪-4-酮平面扭曲约28°(图8)。哒嗪-4-酮母核两个骨架跃迁为两个吡啶-2-酮的变体得到生物等排体化合物27,其对PDE10A的抑制作用仅比26低3倍,是活性衍生物25的300倍。当与PDE10A结合时,结合模式无法解释为什么25的活性下降,我们怀疑配体的张力能是潜在原因。量子力学(QM)计算确实揭示了两个杂环之间扭转角τ的能量分布存在重大差异。虽然26和27(红色和蓝色曲线)的最小能量与蛋白质结合构象中的扭转角很好地吻合,但25的优势构象却扭曲得多(品红色曲线,在τ≈90°时最小)。在τ≈30°处有相当大的配体张力能(2-3 kcal/mol),这将转化为结合亲和力下降约100倍。随后合成的化合物证实了这一观察结果。例如,吡嗪-2-酮化合物28的二面角极小值在τ= 0°处,但该化合物仍然有效,这是因为在τ= 30°时,配体张力受到限制(0.3 kcal/mol)。由于计算的配体张力能与IC50值之间具有良好的定量一致性,因此我们进行了类似的二面能量扫描,以便优先考虑虚拟杂芳基的骨架修饰,然后再开始合成(结果未显示)。
当实验结构信息不可用时,同源模型在多大程度上也可以使用?下面的两个例子说明了即使是相对粗糙的蛋白模型也可以为项目指出明确的方向。成功的关键是充分利用同类靶标的机理方面知识,加上比在实验确定的结构更低的粒度水平上所解释模型的理解。
Hormone sensitive lipase (HSL)
激素敏感性脂肪酶(HSL)在储存脂肪的动员中起关键作用。糖尿病患者由于该脂肪酶的上调而表现出增强的脂解活性。 HSL抑制被认为能恢复过高的血浆游离脂肪酸和甘油三酯水平,因此对治疗2型糖尿病具有治疗价值42。
在项目开始时,所有已知的HSL抑制剂都是自杀性底物,对靶标产生不可逆抑制。高通量筛选发现了磺酰哌啶化合物29,这是第一种具有弱的、但可重现的细胞活性的可逆抑制剂(图9)。化合物29的活性可通过酰胺键环化得到螺内酰胺哌啶的30而显著提高。该化合物的初步小鼠研究表明,当以30 mg/kg的剂量给药时,游离脂肪酸可减少了21%。螺内酰胺哌啶系列化合物具有低溶解度、低代谢稳定性的缺点,因此药物化学团队开始着手优化这些性质。

Figure 9.(a)对HSL高通量筛选苗头化合物29酰胺键的环合发现了化合物30。(b)化合物30与HSL同源模型结合位点的理论结合模式。(c)化合物31与同样的HSL模型的理论结合模式。(d)化合物31是将化合物30的磺酰基哌嗪替换为环己醇而设计化出来的。非对映异构体32的活性损失进一步支持了该理论结合模式。
为了研究理论结合模式能否用来指导优化过程,建立了HSL的同源性模型。 HSL与实验确定的3D结构的蛋白质几乎没有序列同源性。所使用的三个模板与HSL的整体序列一致性仅为12-14%,序列相似性为22-24%。但是,由于包埋的活性位点通道具有不同的形状,并且由于初始先导化合物30具有同样的不同形状,因此所得的HSL模型仍使我们能够建立近似的理论结合模式:简单的力场计算可以确定在极性环境下,内酰胺羰基团偏向于伪平伏键的位置,使其更暴露于环境。在该取向下,磺酰胺部分仅具有一个优选的取向,使末端苯环指向羰基的相反方向。在活性位点内,内酰胺羰基与氧阴离子孔很好的契合,将三氟甲氧基指向狭窄的亲脂性口袋。因此,磺酰胺取代基位于活性位点隧道的另一端,在更宽的亲脂性区域中。虽然这种结合假设只是一个粗略的草图,但它为内酰胺部分的作用提供了理论依据,并暗示在磺酰胺位点可能存在更大的变化空间。但是,团队最需要的是给出先导化合物的一个位置以增加极性基团。磺酰胺是个分子嵌合体(chimera),可形成氢键并与蛋白质内的单极性环境相互作用6。在本案例中,同源模型表明磺酰胺氧原子可与氧阴离子孔附近的谷氨酰胺侧链相互作用。我们假设该侧链的酰胺末端同样可以很好地充当氢键受体。因此,我们设计了一种分子,该分子在直立键方向用带有羟基的环己基环取代了磺酰基哌啶、在平伏键位置有个丁基从而占据了亲脂性口袋。实际上,化合物31显示出与30相似的抑制活性,更少的重原子数,因此显著地改善了配体效率。相应的对映异构体32的活性显著损失也证明了该假设的有效性。对于没有丁基的一对化合物,以衰减形式观察到相同的差异活性(数据未显示)。
该实例说明了尽管缺乏可靠的信息、有关酶学机理的、蛋白质结构和构象等先验知识,但还可以激发创造力以建立具体的、可检验的理论模型(假设)。显然,这样的假设并不总是正确的,但是实验设计的越好, 就可以越多地了解需要修改假设的地方。在HSL的情况下,同源性模型没有被篡改,这并不意味着它是正确的,而是它的基本特征与实验数据一致。 最重要的是,项目团队受益于磺酰胺的替换,这在新的方向上扩展了化学空间,并能够创造出具有理化性质均衡且稳定、有效的化合物43。
G-Protein-Coupled Bile Acid Receptor 1 (GPBAR1)
GPBAR1被胆汁酸(如胆酸)激活。它在葡萄糖稳态中发挥作用,并通过Kupffer细胞触发抗炎活性44。GPBAR1是A类GPCR,与视紫红质一样,与Gα偶联。 GPBAR1项目的起点是高通量筛选命中的许多有效的GPBAR1激动剂,它们具有高亲脂性和低溶解性45。图10给出了一种代表性化合物(化合物33,R = H)。由于GPBAR1跟石胆酸(LCA)以及牛磺酸共轭物牛磺胆酸(TLCA)一样的结合良好,因此该受体明显耐受天然激动剂上的极性或阴离子侧链,并由此产生了在当前系列化合物的何处引入极性基团的问题。为了指导药物化学工作,需要建立有充分根据的结合理论模型。由于GPBAR1与其他A类GPCR的高度同源,因此可以建立同源性模型(视紫红质模板,PDB 1F88)。然而,将基本无特征的亲脂性化合物对接入跨膜结合口袋并不是一项简单的工作。在没有其他信息的情况下,可能会出现几种不同的取向,每种取向都暗示了一种用于添加极性基团的出口载体(exit vector)。
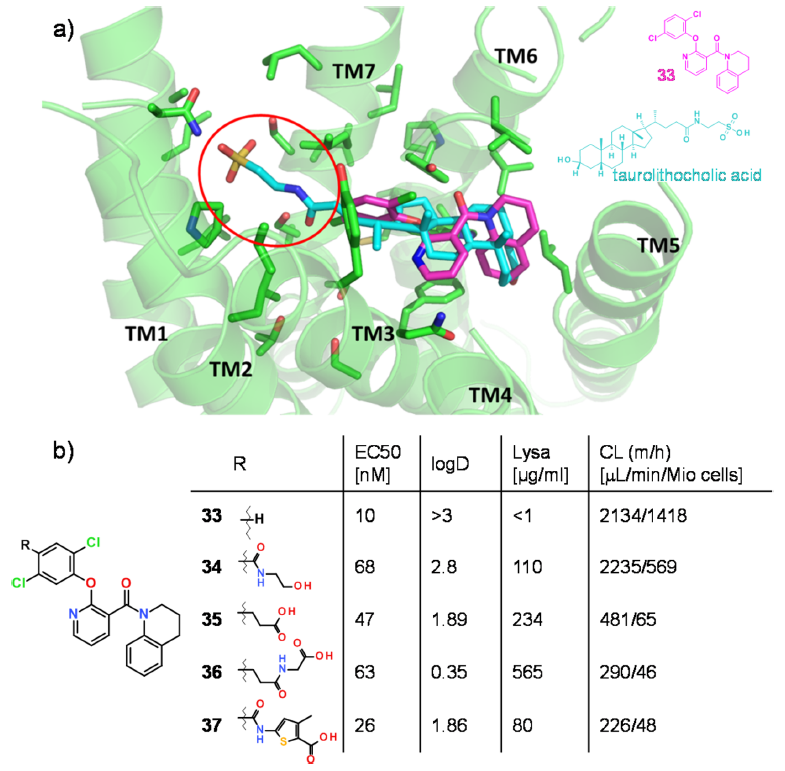
Figure 10.(a)GPBAR1同源模型与TCLA(蓝绿色)以及激动剂代表性化合物(品红色,33,R = H)理论结合模式。牛磺酸片段结合的区域用红色圆圈标记。(b)吡啶系列化合物的GPBAR1活性、亲脂性(log D)、动力学溶解度(LYSA)和微粒体清除率(CL)。
缺少的信息来自该团队正在处理激动剂的事实,该激动剂的分子作用机制可以与A类GPCR视紫红质进行比较。在视紫红质中,视网膜的β-紫罗兰酮挤压在跨膜螺旋5和6(TM5和TM6)之间,这两个螺旋相对于彼此的方向产生小的位移接着转化为TM6在受体胞质侧向外旋转出更大空间,从而允许其容纳G蛋白。因此,在正构结合口袋中,TM5和TM6之间的间隙被认为是同样重要的激动热点。因此,胆汁酸和TLCA以这样的方式对接到GPBAR1模型中,即该热点被类似于视紫红质中 β-紫罗兰酮环的亲脂性部分占据。对于这种结合模式,代表性的先导化合物可以通过保留与激动剂热点的结合方式进行叠合。这导致了单个可能的结合取向,如图10a所示。最有希望的极性侧链出口载体是苯环的对位。如图10b所示,具有各种侧链的化合物确实是强效的激动剂,其溶解度和体外清除率值大大提高。
GPBAR1的案例再次说明,在没有实验确定的靶标蛋白结构的情况下,各种来源的知识(本文为天然配体和对靶标类别的机理理解)的组合也可以生成靶向假设。 此处,对可能出口载体的识别高效地将药物化学搜索空间缩小到特别有希望的区域。
在下一个示例中,我们将离开3D靶标结构的领域。在它们不存在的情况下,生物活性构象的陈述当然会变得更加模糊,因此必须更加谨慎。不同化合物类别的3D叠合和3D药效团生成是有用的技术,其结果当然会受到输入构象质量的强烈影响。但是,配体叠合几乎总是会产生高度3D相似度的错误外观。不同结构类别的结合模式彼此间的差异通常远大于叠合所建议的那样。这个问题困扰了3D-QSAR领域很长一段时间,因为当结构多样性低或它们很可能出错时,叠合就不是什么问题。在这种情况下,生成可直接测试生物活性构象假设的刚性结构是显而易见的。默克Orexin项目是此类验证实验的一个很好的例子46。下一个例子展示了配体叠合如何在不被过度解释的情况下用来指导药物化学。
Liver Carnitine Palmitoyltransferase 1
肉毒碱棕榈酰转移酶是一种线粒体酶,负责将长链脂肪酰基CoA的酰基向L-肉毒碱转移,促进脂肪酸在线粒体膜上的转运47。本项目的目标是识别肝脏同功酶(L-CPT1)抑制剂,其选择性高于肌肉同功酶(M-CPT1)以及线粒体膜上催化脱乙酰化反应的酶CPT2。2004年,对LCPT1进行了高通量筛选。对第一轮命中的5000个化合物进一步用三种酶进行了8点IC50表征,获得约2200个苗头化合物,IC50值低于50 μM,包含多个易处理的苗头化合物类别。在项目的早期阶段,CADD的作用是什么? 基于子结构的聚类(MCS,MOS)[48]和手动叠合可以确定大多数苗头化合物可能以其生物活性构象摆出相同的形状(图11a)。由于推定的生物活性形状“外皮”是手性的,但苗头化合物结构不是手性的,在没有其它信息的情况下,无法区分两种可能的对映异构体形状“外皮”(图11b)。L-CPT1的同源性模型可以进行区分:化合物太大而不能放入适合酰基肉毒碱结合的隧道,因此假定它们将在CoA结合位点结合。该结合位点仅适合两个对映异构体中的一个。在探索苗头化合物的初始阶段,使用图11b中所示的形状外皮来对苗头化合物的合成进行优先级排序,并将不同苗头化合物的元素彼此混合和匹配。D-脯氨酸衍生物39(而不是其对映异构体)应该是L-CPT1抑制剂的正确预测支持了生物活性形状假设(图11c)。
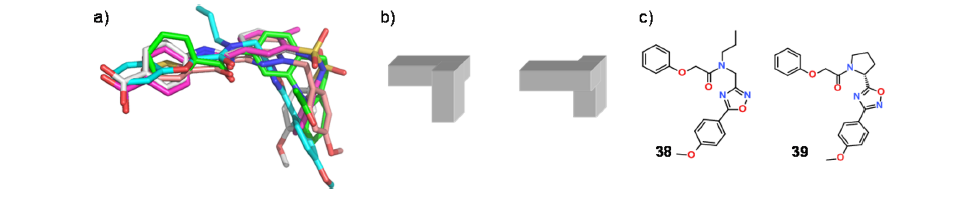
Figure 11.(a)主要苗头化合物类别的代表性化合物的手动3D叠合。(b)两个对映体生物活性形状外皮的示意图,其中左边的那个认为是正确的。(c)化合物39由高通量筛选苗头化合物38衍生而来。用形状外皮假设正确地预测出39对映异构体为活性化合物。
一旦项目团队对多种苗头化合物完成探索,药物化学便迅速专注于优化三个有前景系列的选择性、溶解度和稳定性。 从这时开始,该项目本质上是一个经典的药物化学优化项目,其中的预测方法侧重于分子性质,而不是结合模式和亲和力。
可以得出结论,形状分析有助于发现配体之间的相似性,有助于在项目团队内启动配体片段的”混合和匹配”的尝试。叠合是逐步的、手动处理的过程,其不可能很容易地自动化。其次,没进行简单的形状分析,就不要持续优化结合模型是一个很好的决定。任何定量的3D药效团假设,都可能是基于多个配体的重叠而建立的,可能会将团队带入歧途。迄今尚未获得L-CPT1结合假设的实验验证。然而,我们能够用解释CPT2与非选择性抑制剂的晶体结构,与简单的叠合相比,CPT2确实显示出更宽范围的结合模式49。
缺乏可利用的配体或蛋白质3D信息标志着从经典分子设计向通常被称为”化学信息学”的转变。这一领域的许多方法已经如此深入到日常工作中,以至于它们的假设或设计特征不再被实现:简单的相似性算法通常用于聚类、HTS跟踪、集中库筛选数据库的选择或专利分析50,51。 化学信息学技术与通用的信息科学和生物信息学密切相关。如果将各种不同来源的信息进行交叉链接、提供上下文和降低数据访问障碍的方式来实现它们,则它们是最有效的。下面的最后一个例子说明了这种思维方式52。
SST5R
生长抑素(SST)是一种环状四肽激素,主要抑制荷尔蒙分泌,如生长激素、胰腺胰岛素、胰高血糖素和胃泌素的释放53。SST通过五个不同的G蛋白偶联受体(GPCR)起作用。 当我们寻找SST受体亚型5(SST5R)的选择性抑制剂作为潜在的抗糖尿病药时,尚无已知的小分子拮抗剂。 由于我们不想投入大量资源进行高通量筛选,而基于结构的设计也遥不可及,因此,集中库筛选似乎是快速识别化合物的唯一可行途径。
集中库筛选需要种子化合物进行相似性搜索。 我们没有遵循通常的方法在SST中寻找具有活性四肽的拟肽54,而是基于生物学相似性选择了另一种方法。 根据hSST5受体跨膜结合口袋内氨基酸的相似性,对具有已知小分子配体的A类GPCR进行了排序。仅考虑了共有结合位点周围的局部序列相似性,这种方法在比较GPCR结构时可显着提高分辨率55。
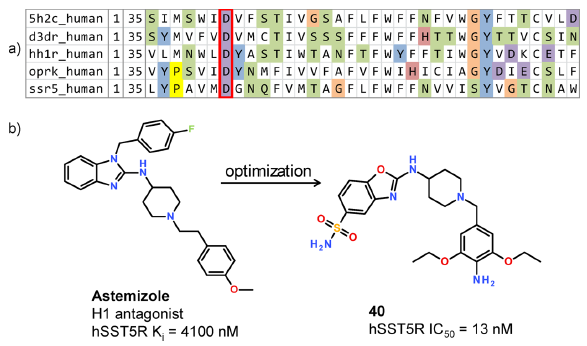
Figure 12.(a)分析GPCR中共有药物结合位点周围的氨基酸同源性。 生物胺受体(血清素、多巴胺、组胺、阿片类药物)的跨膜结合口袋与hSST5R密切相关。高度保守的天冬氨酸(D)用红色框标记,据信阿司咪唑的哌啶环中的叔胺与之非常接近。(b)从阿司咪唑到化合物40,第一个类药的高选择性SSTR5拮抗剂小分子。
SST5R的最近邻居被确定为生物胺受体,通常将其碱性的胺靠近到跨膜配体口袋中保守的Asp332。因此,我们选择了阿片类药物、组胺、多巴胺和5-羟色胺受体的配体对SSTR5进行测试(图12a)。hSST5R放射性配基结合试验识别了许多具有微摩尔亲和力的苗头化合物。从这些苗头化合物中,我们选择了第二代抗组胺药阿司咪唑作为进一步优化的起点。
药物化学团队随后系统地研究了先导化合物的SAR,并设法从苯并咪唑核心去除苄基来降低阿司咪唑的组胺H1亲和力。以这种方式产生了对其他SST受体亚型具有高选择性的、强效和代谢稳定的hSST5R拮抗剂(图12b)56。总之,该实例表明,定制的相似性计算方法与已知配体数据库的系统搜索相结合可以有效地发现不明显的化学进入点。这项工作的先决条件是适当的数据基础:带注释的配数据集以及正确比对的跨膜口袋序列。
结论
我们相信,这10个罗氏案例研究虽然并不详尽,但可以代表当今分子设计可以为小分子药物发现提供的技术。 具体的工具和方法对许多类型的项目都具有广泛的用途,包括我们此处未介绍的项目:基于片段的药物设计,蛋白质-蛋白质相互作用抑制剂的设计,变构调节剂等。 分子识别的基本规则保持不变,而难度和可预测性却有所不同。 我们现在要后退一步,找到这里使用的各种工具和方法背后的共同主题。
1. 定性陈述的价值
通常,对于项目团队而言,一个新的想法或指向新方向的指导就足够了。 大多数项目影响来自于定性工作、分享见解或假设、而不是计算得出的数字或优先级顺序。在一个对定量预测方法投入巨大的领域,这种观察的重要性怎么评估都不过分。 我们认为,仅仅用定量预测是对分子设计使命的误导性陈述。 计算工具就其本质而言,当然可以产生数值结果,但是绝对不能如此使用。 相反,任何排序列表都应被视为原始输入,以便在项目范围内进行进一步评估。 例如,在性质预测中选择分类而不是回归模型,该原理可以被非常广泛地应用,并且超出结合亲和力预测的问题。
2. 塑造化学空间
药物化学是一门创造的学科。在项目进行过程中的任何时候,团队的重点要么是扩大化学空间,要么是缩小化学空间,以解决和优化问题的不同方面。扩大化学空间需要在一系列约束条件下产生新想法的方法。在这种情况下,有用的工具是基于查询的工具:3D骨架跳跃,药效团或形状搜索的工具,或将已知化合物的元素相互重组的工具。缩小化学空间可以是简单的过滤过程,也可以基于特定的假设(如上述GPBAR1案例)。在给定的项目环境中,重要的是要了解是否需要扩大或缩小化学空间并相应地选择工具和方法(参见图1的图例)。随着项目向候选药物选择发展,缩小和扩大空间的“幅度”通常会变小,但概念保持不变。先进的先导优化中的理性方法可能,例如,建议避免使用某种子结构或优先使用某种取代模式以避免脱靶效应或改善ADMET特性。
3. 简单即好的原则
分子设计是一个概念过程,因此始终有失去与现实联系的风险。 科学问题应引出方法,反之亦然。 为此,使事情尽可能简单是一个有用的指导原则。 选择最简单的解释和最简单的计算流程可以提高灵活性,并更好地关注手头的关键问题。 一个典型的例子是在任何可能的情况下使用从实验获得的数据(例如,构象的CSD),并仅在需要时(例如,杂环之间的扭转角或互变异构体偏好)诉诸第一性原理方法。
4. 注释是成功的一半
上述组织蛋白酶示例说明了如何用正交视角看到设计机会。对复合物晶体结构可视化分析其中有利与不利的相互作用、水分子形成的接触、柔性区域等信息来会带来价值。上下文信息几乎可以在任何地方带来增值。通常需要进行大量的前期工作(计算,组织)来将数据成转化为有用的形状。 正确的前端加载工作可以将复杂的查询转变为简单的查找过程或可视化步骤。 除了结合模式可视化之外的良好实例是通过匹配分子对来重复使用ADMET数据或者上述针对SST5量身定制的生物信息学方法。 这方面有很大的增长潜力57,58。
5. 与实验近距离
使事情尽可能简单的一种方法是优先利用可能支持项目的实验数据,只要这是有意义的。这可以通过许多不同的方式来完成:参考测得的参数而不是计算的参数;利用现有的化学砌块而不是设计新的化学砌块;充分利用已知的配体和SAR或相关的蛋白质结构。合理的药物设计与巧妙的回收利用有很大关系。
如果得到贯彻,这些指南会重新把务实的重点拉回到当前的分子设计实践。 让我们看看在目前实践中存在的一些问题。
许多计算方法引入额外的参数并因此引入潜在的误差源,这使得预测值难以提取。处理蛋白质动力学的方法属于这一类别。在大多数基于结构的项目中,可以利用刚性蛋白质结构或利用侧链的选择性松弛或旋转来建立粗略的结合假设或能量估计。自由能微扰方法也有类似的论点。这一领域取得了明显的进展59-61。然而,在定性答案就足够好的快节奏项目中,节约原则限制了它们的使用,因为它们所带来的额外复杂性收效甚微。此外,它们的适用范围小,因为水结构或构象的显著变化难以解释和量化。
在需要评估蛋白质动态性质(Mobility)且无法通过其他技术进行评估的情况下,分子动力学(MD)可能会有用62,63。需要格外小心,不要过度解读或曲解计算结果。MD轨迹无法通过实验验证,因此需要付出额外的努力才能将此类模拟结果与真正可检验的假设联系起来,例如,在定性预测机制或蛋白质运动可能用于结合剂(binder)设计的场合。
近年来,已提出了尝试在蛋白质结合位点内模拟水网络的方法64-66。在此,该方法的直接计算结果(水位,结合能,熵变)无法通过实验进行测试,因此, 将结果转换为会推动药物发现的假设,而不是提供需要额外的努力事后合理化。这额外的努力是什么?至少,应该弄清楚底线是相对于哪些计算是一种改进。 就水的结构和动力学而言,这可能是经典的去溶剂化观点或晶体学或模拟水位置的几何打分67。如果没有这样的参考模型,从更高的理论水平上计算水的作用,几乎没有机会获得新的见解。
每种方法都有其适用范围和一组必须避免的内在缺陷。如上所述,最典型之一是对数值结果的过高置信度。例如,小分子-蛋白对接可用于确定化合物的优先级以进行实验筛选。对接程序可以评估化合物是否以合理的构象与结合口袋匹配,但众所周知,对接打分值与结合亲和力数据无关68-71。正确使用对接程序需要使用尽可能多的约束条件,并在视觉检查和选择方面投入大量资金72。如果没有进一步的审查,对接工具预测的结合模式不应被接受。只要可能,药效团搜索或形状匹配提供更集中假设的测试73,74。
比较不同的配体时,基于特征的叠合结果通常给人以高3D相似性的印象,而这一点并尚未被实验证实。 结合模式通常大大地有别于3D相似性所暗示的。无论何时进行基于此类叠合的预测时,都应牢记这一点。因此,只有对生物活性构象作出明确的陈述时,才能有把握地推导出3D药效团和形状约束。对于大多数QSAR方法,存在生成自洽模型的风险,该模型本质上反映了手头的数据,但没有预测能力。 从药物化学家的角度来看,它们还有另一个缺点,那就是黑匣子,只能用作过滤器,而不能培养创造性思维。
分子设计的最佳实践是所有科学的最佳实践:对清晰、简单和良好实验设计的不懈关注。 分子设计的特殊之处在于需要建立扎实的假设并同时培养药物化学方面的创新思维。 如果我们接受这一点,我们的重点可能会从我们拥有的许多半定量预测工具转移到支持这一创造性过程的方法上75。那么,计算方法的进一步改进可能与科学的关系不如与好的软件工程和界面设计的关系。 这些工具只是达到目的的一种手段。 如果适当地使用它们,就会产生好的科学。


