
Gaussian教程 | Michael受体与巯基模型的加成反应和反应速率常数的计算
摘要:我们选取了四个Michael受体,分别计算了这些受体与巯基模型的加成反应、计算了能垒并构建了反应势能面。同时我们也介绍了如何运用过渡态理论中的Eyring公式计算反应的速率常数。这为基于Michael受体与Cys反应的共价抑制剂设计提供了理论指导。
作者:陈宇
时间:2020-03-28
前言
对比传统的非共价抑制剂,共价抑制剂可实现更高的结合亲和力和更长的停留时间,这些特性可以变现为更低的剂量、给药频次和药物的全身性暴露。因此,许多制药公司启动了针对各种酶的共价抑制剂药物开发计划。其中一个策略是“靶向共价抑制”,该策略是把共价的弹头与非共价相互作用识别靶蛋白结合位点的配体相捆绑。一旦配体对接进入结合位点,弹头就会与附近的氨基酸残基形成共价键。通过设计抑制剂使共价键仅形成于保守性差的非催化残基,以此实现靶标选择性的最大化。
共价抑制剂与蛋白结合过程分两步:(1)首先形成可逆的初始非共价复合物EI;(2)初始的复合物EI进一步过渡为共价键结合的复合物E-I:
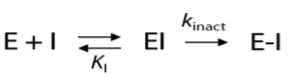
共价抑制剂的酶学活性用二级速率常数kinact/Ki来表示。kinact反映了共价键的形成速率,而Ki反映了通过非共价相互作用形成初始复合物EI的稳定性。第一步过程可以用基于结构的方法(比如分子对接)进行模拟;第二步涉及键的生成与断裂,需要用QM的方法进行模拟。我们在《用常规分子对接进行共价抑制剂的虚拟筛选》 一文中已经详细地讨论过第一步的模拟,而本文主要讨论第二步共价键的形成。
在之前的教程中[1],我们已经计算了四种Michael受体与甲胺的加成反应。由于Michael受体的半胱氨酸巯基加成是靶向共价抑制剂设计的常见形式,因此在这个教程中,我们将研究几种Michael受体与甲硫醇的加成反应。
利用Gaussian程序进行过渡态搜索
在这里,我们选择了四个Michael受体,同时用甲硫醇作为巯基模型,计算了下述四个反应过程(图1)的能垒,其中机理的猜测主要依据KN Houk的研究工作[2]。首先,甲硫醇在碱的作用下生成甲硫负离子,然后甲硫负离子进攻Michael受体,并最终得到加成产物。
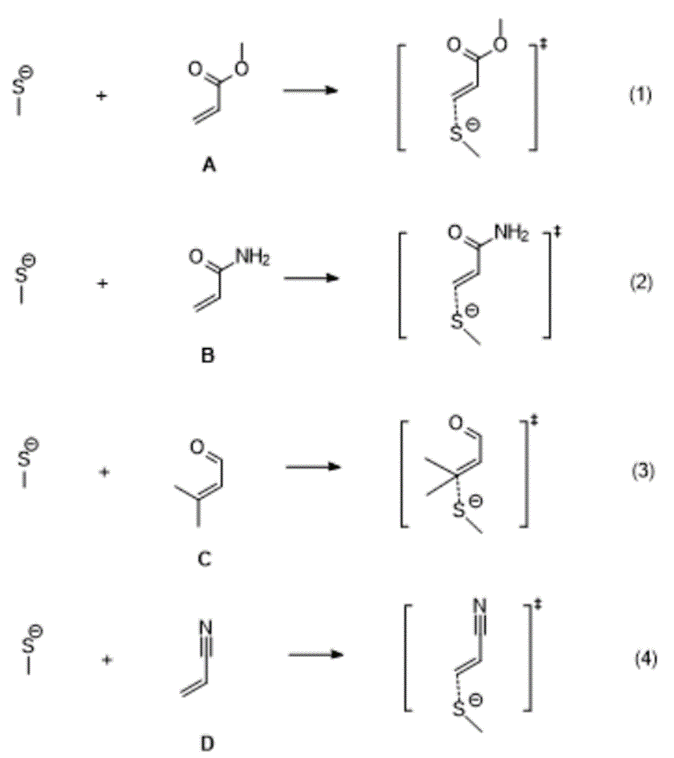
图1. 四个Michael受体与CH3SH的反应过程
过渡态的成功搜索依赖于能够构建一个合理的过渡态结构的初始猜测,许多方法能够达到这一目的,在这里我们为寻找简单体系的过渡态提供一个简单通用的流程:
1. 优化反应物
首先,优化参与到过渡态的反应物分子的结构,这个例子中包括Michael受体A和甲硫负离子。
优化分子A的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | %nprocshared=24 %mem=1GB # b3lyp/def2tzvpp opt freq scrf=(iefpcm,solvent=water) em=gd3bj A 0 1 C -1.62628800 -0.07029800 -0.12431700 O -1.60403200 0.02468800 -1.37556100 C -0.67047100 -0.75005600 0.67050000 H -0.76922500 -0.69699000 1.74815000 C 0.45266800 -1.38578500 0.10261400 H 0.36572300 -1.64470600 -0.94347400 H 0.93850600 -2.15192900 0.69188300 O -2.68826586 0.58988251 0.56941546 C -3.62741005 1.10733150 -0.37671071 H -4.54071239 1.35162817 0.12438607 H -3.22510775 1.98708530 -0.83397006 H -3.81912601 0.37046295 -1.12848842 !空白行都是特意留出,不要删除 |
其中scrf关键词表示在能量计算中考虑溶剂化作用;em=gd3bj表示在能量计算中添加色散校正。
优化甲硫负离子的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | %nprocshared=24 %mem=1GB # b3lyp/def2tzvpp opt freq scrf=(iefpcm,solvent=water) em=gd3bj MeS-anion -1 1 C 1.15997500 0.01968500 0.00001100 H 1.51839200 0.52399000 0.89284000 H 1.52419800 -1.00480800 -0.00191300 H 1.51843600 0.52721000 -0.89096400 S -0.66284700 -0.08718900 0.00000000 !空白行都是特意留出,不要删除 |
2. 优化过渡态结构的初始猜
首先,将优化好的两个反应物分子复制到同一个分子构建界面中,由于加成反应是甲硫负离子的S原子进攻Michael受体的双键,因此我们将S原子置于双键Π轨道的位置,然后调整C和S原子的距离,最理想的情况是已经有文献报道这种类型的过渡态结构,我们可以直接调整到相同的距离;若是没有文献可参考,我们可以大致设置一个距离,但一定要比产物的C-S键距离大,然后根据优化结果在重新调整距离,在这里我们设定C-S距离为2.2埃。
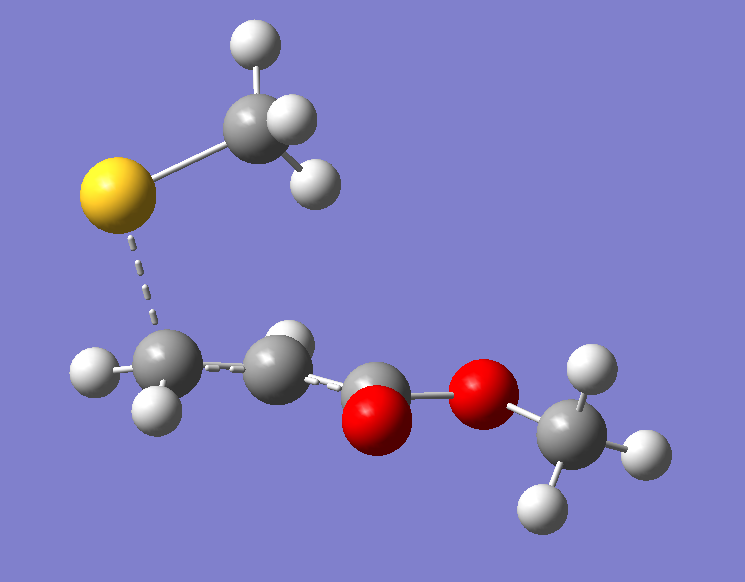
图2. 过渡态结构的初始猜
接下来,保存好结构,编辑输入文件,即可提交文件开始过渡态的寻找,在这个例子中,只进行了一次初始构型猜测便得到了过渡态结构。对于更复杂的过渡态结构,我们可能很难一次得到过渡态结构,这时需要根据失败的结果调整初始构型,使其更靠近真实的过渡态结构,以增大成功的概率。
过渡态优化的输入文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | %nprocshared=24 %mem=10GB # b3lyp/def2tzvpp opt=(calcfc,ts,noeigen) freq scrf=(iefpcm,solvent=water) em=gd3bj TS-A-anion -1 1 C -1.62628800 -0.07029800 -0.12431700 O -1.60403200 0.02468800 -1.37556100 C -0.67047100 -0.75005600 0.67050000 H -0.76922500 -0.69699000 1.74815000 C 0.45266800 -1.38578500 0.10261400 H 0.36572300 -1.64470600 -0.94347400 H 0.93850600 -2.15192900 0.69188300 C 1.45981700 1.49469500 0.16419900 H 0.44819800 1.30918600 0.54579000 H 2.00791200 2.08183100 0.89960700 H 1.38562200 2.06348200 -0.76218500 S 2.23928400 -0.11847300 -0.08898600 O -2.68826586 0.58988251 0.56941546 C -3.62741005 1.10733150 -0.37671071 H -4.54071239 1.35162817 0.12438607 H -3.22510775 1.98708530 -0.83397006 H -3.81912601 0.37046295 -1.12848842 !空白行都是特意留出,不要删除 |
3. 检查优化的结构是否为需要的过渡态结构
首先查看是否只有一个虚频,如果只有一个虚频,说明优化的结构是一个过渡态结构,但这并不保证是我们想要的过渡态结构,因此需要进行一个IRC计算,以此来确定这个过渡态连接反应物和产物。
详细的计算过程见:Gaussian教程 | 搜索过渡态。
4. 提取自由能
当我们优化完所有的结构后,需要构建反应势能面,这其中就包含了反应活化能的信息,我们需要提取每个分子的自由能信息。我们以分子A加成反应的过渡态为例,用文本编辑器打开log文件,找到下面的信息,最后一行(第8行)即为自由能。
1 2 3 4 5 6 7 8 | Zero-point correction= 0.132122 (Hartree/Particle) Thermal correction to Energy= 0.142074 Thermal correction to Enthalpy= 0.143018 Thermal correction to Gibbs Free Energy= 0.095305 Sum of electronic and zero-point Energies= -744.766315 Sum of electronic and thermal Energies= -744.756364 Sum of electronic and thermal Enthalpies= -744.755420 Sum of electronic and thermal Free Energies= -744.803132 |
结果分析
1.构建反应势能面
Michael A与Cys模型分子CH3SH反应的活化能(反应能垒)根据下式计算:
Reaction Energy Barrier, ΔG = G‡ – (GA + GCH3SH)
= -744.803132-(-306.555707-438.264567) Hartree
= 10.8 kcal/mol
利用同样的方法我们计算出了4个Michael受体与巯基模型的过渡态,并构建了加成反应的势能面(图3)。若要得到完成的势能面,读者可以尝试优化加成产物的结构,取能量最低的构象,然后提取分子的自由能,减去两个反应物自由能的加和,再换算成kcal/mol,最后标注在势能面上即可。
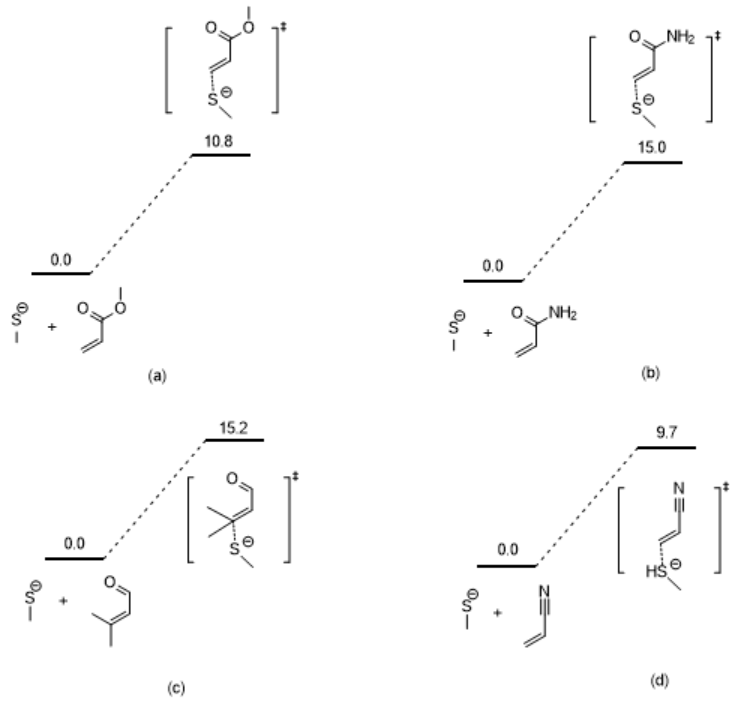
图3. 四个Michael受体与甲硫醇反应的反应势能面
需要注意的是:在方法基组选择合适的情况下,活化能计算的准确性依赖于构象的选择,因此得到能量最低的构象非常关键,如果构象不合适,甚至会得出错误的结论。
2. 计算反应速率常数
接下来,我们介绍如何计算反应速率常数。在过渡态理论中,Eyring公式给出了反应速率常数的表达式:
$$
k(T) = \frac{K_B T}{hc^0} \times e^\frac{-\Delta G}{RT}
$$
其中k(T)是指温度为T时的反应速率常数,kB为玻尔兹曼常数,h为普朗克常数,c0为标准浓度(通常取为1),R为理想气体常数,ΔG为计算的反应活化能垒。
我们以上面图6(a) 的反应为例,其活化能为10.8 kcal/mol,同时我们是在298.15K下计算的活化能,因此代入上述公式:
$$
k(298) = \frac{1.389662 \times 10^{-23} \times 298.15}{6.626176 \times 10^{-34} \times 1} \times e^{(-10.8 \times 1000)/(1.987 \times 298.15)}
= 7.516 \times 10^4 s^{-1}
$$
相关内容
- ONIOM模拟
- QM/MM计算
本文以甲硫醇作为酶-Cys的模型分子来模拟Michael受体与Cys的共价结合过程,这与实际的酶-Cys与Michael受体的加成反应还差异很大。Guassian的ONIOM可以更加准确的模拟酶与Michael受体的反应,详细的计算过程请查阅《探索化学的奥秘:电子结构方法》第三版第397页的第9章高级模拟技术。
Gaussian与AMBER联用的QM/MM是一种成熟的技术,可以用来模拟酶与底物的反应,计算反应历程与能垒,更详细的内容请参见:培训通知 | 用QM/MM模拟酶与底物的反应。
文献
- 陈宇, 肖高铿. Gaussian教程 | 用DFT预测Michael受体的遗传毒性(AMES test). http://blog.molcalx.com.cn/2020/01/08/dft-predict-ames-test.html
- Krenske EH, Petter RC, Houk KN. Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J Org Chem. 2016;81(23):11726-11733. doi:10.1021/acs.joc.6b02188


