
摘要:本教程为提供了Linux下Gaussian 16的安装流程:1) 指令集的检查与确认;2)Gaussian的安装;3)Gaussian 16的环境变量的设置。适用于没有Linux基础的同学学习如何在Linux下安装Gaussian软件,提交作业。
肖高铿 2019-12-13 修改
一. Gaussian 16的安装
强烈建议请参考Gaussian Inc官方的安装教程。首次购买Gaussian的用户,在收到光盘的袋子里有也附有一份安装指南,请仔细阅读。
因为G16 Windows版的安装方法与其它的Windows软件的安装方法没有任何区别,所以本文仅举例说明如何在Linux下安装。
因此您需要具备必要的Linux基础知识。请参考《Linux命令极简教程》。我个人认为梁如军,王宇昕,李亚军等编著的《Linux基础及应用教程(基于Centos 7)》(机械工业出版社)不错,微信读书里等有电子书可购买。您只要花半天或一天的时间去学习,将会从Linux获益无穷。如果您不愿意学习linux,本文就没有办法帮助到您。对于Linux的初学者,建议您采用英文界面,可以避免很多”照着你的方法,我为什么不能…?”的问题。
本教程假定:
- 采用光盘安装
- 使用bash环境
- 安装软件的用户名为tom
通常,Gaussian 16安装文件以.tbJ结尾,假设安装文件为foo.tbJ。不少同学抱怨不知道如何解压。如果你将文件从Windows机器上传到Linux系统,请不要在Windows下解压安装文件!
假设tom的HOME目录(~目录)为:/home/tom,并计划软件将软件安装在/home/tom目录下。观察到很多人因此用root身份在/home/tom下创建了tom目录,引发了很多问题。在此特意强调:/home/tom这个目录是通过创建用户生成的,而不是单独为了创建这个目录生成的。
关于G16与GaussView 6/Linux同时安装
先安装Gaussian 16,再安装GaussView 6。具体方法参见《GaussView 入门教程》。
关于Linda的安装
Gaussian分带Linda的版本(跨节点并行)与不带Linda的版本(单机多核的共享内存并行,SMP)。带Linda的Gaussian与不带Linda的Gaussian在安装上没有任何区别。
二. 安装流程
第一步:确认自己的操作系统类型与适用版本
Gaussian 16支持INTEL/AMD 64BIT CPU, 还针对SSE4.2,AVX, AVX2指令集做了优化。因此,需要先了解自己的机器支持哪种指令集以便选择合适的Gaussian版本进行安装。请在操作系统shell命令行键入:
1 | grep -o -e sse4_2 -e avx -e sse4a -e avx2 /proc/cpuinfo |
- 如果包含有avx字眼(无论有无sse4_2或sse4a字眼),建议您使用AVX指令集的Gaussian版本。
- 如果什么都不出现或仅有空白行,则您的机器不能支持SSE4.2或AVX指令集,需要使用Legacy版本进行安装;
- 如果包含有sse4_2或sse4a字眼而没有avx字眼,建议您SSE4.2指令集的Gaussian版本;
- 如果包含有avx2字眼,建议您使用支持AVX2指令集的Gaussian版本;
- 如果您的集群既有Legacy又有AVX等机器,为了避免出错,建议您用Legacy版本。
第二步:环境变量$g16root的设定
第三步:切换到$g16root目录,并且解压安装文件
[tom@linuxbox ~]$ tar -zvxf foo.tgz
解压后,在/home/tom目录下会生成一个新的目录:g16。
如果安装文件是.tbz结尾,请将tar的参数由-zvxf改为-jvxf:
如果安装文件是.tbj或tbJ结尾,请将tar的参数由-zvxf改为-Jvxf:
具体的安装方法,在安装光盘的REAME里有详细的描述,请仔细阅读。
第四步:完成安装
[tom@linuxbox g16]$ ./bsd/install
至此,已经完成安装,可以开始使用Gaussian 16了。
第五步:GaussView 6与Gaussian 16一起安装
方法:先安装Gaussian,再安装GaussView。假设GaussView安装文件为gv.tgz,在安装完Gaussian之后,键入如下命令:
[tom@linuxbox ~]$ tar -zxf gv.tgz
总的来说,需要切换到$g16root目录,将GaussView解压到该目录即可。
GV6在Linux系统下的使用详见《GaussView 6教程》。
二. Gaussian 16环境变量的设定
Gaussian 16使用前需设定使用环境,先设定$g16root与$GAUSS_SCRDIR两个环境变量。
1. $g16root与$GAUSS_SCRDIR环境变量的设置
设置环境变量$g16root,建立tmp目录作为scratch目录:
[tom@linuxbox ~]$ mkdir $g16root/tmp
[tom@linuxbox ~]$ export GAUSS_SCRDIR=$g16root/tmp
注意:G16在计算可能会在$GAUSS_SCRDIR里产生很大的临时文件,如果这个变量所在的目录空间不足或读取速度不够快会影响G16的使用,这里只是一个例子,请根据自己的情况灵活设定。
2. 加载g16的其它环境变量:
简便的方法是建立一个文件,比如为g16_env.sh,预先设置好各个所需的环境变量:
1 2 3 4 5 | #-----g16_env.sh Start----- export g16root=~ export GAUSS_SCRDIR=$g16root/tmp . $g16root/g16/bsd/g16.profile #-----g16_env.sh End----- |
每次,在使用高斯或高斯视图(GaussView)前先键入下列命令:
还可以将g16可执行文件目录添加到PATH里,并将下面内容添加到主目录的.bashrc里,实现打开shell可以直接使用Gaussian及其应用程序比如cubegen,cubman,formchk等:
1 2 3 4 5 6 | #-----g16_env.sh Start----- export g16root=~ export GAUSS_SCRDIR=$g16root/tmp . $g16root/g16/bsd/g16.profile export PATH=$PATH:$g16root/g16 #-----g16_env.sh End----- |
注意:要特别注意空格(” “),句点(“.”),主目录(“~”)与美元符(“$”)不要漏写了。
使用cat命令,可以非常方便的将上述环境变量追加写到.bashrc文件里:
退出shell,再重新登陆shell或者重新加载环境变量(用命令:source ~/.bashrc)就可以直接使用g16命令。注意:当你有多个不同的Gaussian版本同时在用的时候,不建议将上述环境变量加到.bashrc里。
更多的如何执行一个Gaussian作业,请参考:http://gaussian.com/man
3. AMD的Zen+/Zen2构架(architecture)用户
Gaussian采用PGI编译,AMD Zen+/Zen2出现于Gaussian 16 B.01发布之后,因此G16 B.01以及之前的版本无法正确识别比它晚出现AMD CPU, 也无法识别Zen+构架的AVX2指令集,导致AVX2指令集的Gaussian无法在Zen+构架下使用,并且报错:
1 2 3 4 5 6 7 8 9 10 11 12 13 | Error during math dispatch processing... Error: Fastmath dispatch table is corrupt /opt/admin_home/g16/l302.exe() [0x152e4af] /opt/admin_home/g16/l302.exe() [0x9e6958] /opt/admin_home/g16/l302.exe() [0x75b148] /opt/admin_home/g16/l302.exe() [0x4dc751] /opt/admin_home/g16/l302.exe() [0x4bf664] /opt/admin_home/g16/l302.exe() [0x414bc9] /opt/admin_home/g16/l302.exe() [0x40f58c] /opt/admin_home/g16/l302.exe() [0x40bd2b] /opt/admin_home/g16/l302.exe() [0x40bc34] /usr/lib64/libc.so.6(__libc_start_main+0xf5) [0x2ad37bafd445] /opt/admin_home/g16/l302.exe() [0x4063a9] |
特征错误是第1、2行提示,此时需要额外设定环境变量以解决这个问题:
1 2 | export PGI_FASTMATH_CPU=sandybridge export OMP_THREAD_LIMIT=256 |
也可以通过升级软件的方式解决该问题。
4. 测试一个Gaussian 16自带的算例
万事具备,现在测试一个Gaussian自带的算例test0001.com。
首先建立一个demo目录
[tom@linuxbox ~]$ mkdir demo
[tom@linuxbox ~]$ cd demo
从测试集里复制算例到当前目录:
执行计算:
如果已经设置了PATH,可以直接键入g16命令:
Gaussian 16如果不设置输出文件的名称,默认为:作业名.log,所以上面的命令于下面的一样:
三. Gaussian 09与Gaussian 16的共存与切换
请将Gaussian 09与Gaussian 16的环境变量分别保存在g09_env.sh与g16_env.sh里,需要使用哪个版本,就启用哪个版本的环境变量。比如,需要使用G09,请键入:
四. 共享安装或集群共享安装
有的课题组希望安装一次,多个用户共享使用,请在单用户安装的基础上额外设置用户群组与分配权限,操作步骤如下:
第一步:群组设置
假设:gaussian群组(group)用户都可以使用gaussian,我们需要先将需要使用Gaussian的用户(tom,kim)预先添加到gaussian群组里。
建立gaussian群组:
[root@linuxbox tom]# groupadd gaussian
将用户tom加入到gaussian群组:
[root@linuxbox tom]# groupmems -g gaussian -a tom
同理,希望另一个用户kim也能使用tom安装的Gaussian的话,需要将kim也添加到gaussian用户组:
[root@linuxbox tom]# groupmems -g gaussian -a kim
第二步:给g16目录添加gaussian群组用户的权限
[root@linuxbox tom]# chgrp -R gaussian g16
有时存在未知的原因,Gaussian安装目录g16的所有者在系统内并不存在,需要重新分配用户与组,可以使用chown命令,格式如下:
1 | chown -R 用户名:组名 目录 |
具体的,比如将g16所有者修改为root,组修改为gaussian,则键入如下命令:
[root@linuxbox tom]# chown -R root:gaussian g16
第三步:用户同步(仅限集群里安装)
集群安装最常见的问题是权限设置不成功,主要体现为:单节点组用户执行没问题,但在另一个计算节点上组用户计算失败,不能并行或没有读、执行的权限。请系统管理员做一个用户数据同步即可,因为新增的用户属性需要同步之后计算节点才能知道。一般可以使用下面命令:
[root@linuxbox tom]# make
五. PBS作业管理系统提交作业
1. PBS作业脚本
假设:用户zdtest在其HOME目录的demo里(/hpme/zdtest/demo)提交一个单节点32核计算作业test0397.com, 现在要写一个PBS作业脚本文件为Gaussian计算获取单节点、32核的计算资源。
解决方法:写一个test0397.pbs作业脚本,内容如下:
1 2 3 4 5 6 7 8 | #--------从这里开始-------- #PBS -o /home/zdtest/demo/test0397_pbs.log #PBS -e /home/zdtest/demo/test0397_pbs.err #PBS -l nodes=1:ppn=32 source /home/zdtest/g16_env.sh cd $PBS_O_WORKDIR $g16root/g16/g16 -p=32 -m=64GB test0397.com #-------到这里结束-------- |
2. 提交PBS作业
3. 作业依赖
比如,我希望一个作业等待另一个作业结束之后再启动计算。典型的计算场景:(1)先Opt freq计算;(2)再进行激发态计算。那么需要使用作业依赖,用-W选项:
1 | qsub -W depend=afterok:work1_id work2.pbs |
其中work1_id为第一个作业的作业号, work2会在work1完成之后再自动启动计算。
4. 作业取消
比如,我希望将一个运行中的作业取消:
1 | qdel <作业号> |
六. LSF作业管理系统提交作业
1. 命令行提交作业
(1) 单线程作业
提交一个单线程g16计算作业(test0001.com)到默认队列:
(2) 多线程作业
提交一个32线程g16计算作业(test0397.com)到默认队列:
(3)命令行提交作业的小结
LSF bsub格式如下:
1 | bsub -n Z -q QUEUENAME -i INPUTFILE -o OUTPUTFILE COMMAND |
-n Z: 提交作业需要的线程数;
-q QUEUENAME: 指定作业提交的队列。如果不添加 -q选项,系统将把作业提交到默认的作业队列。
-i INPUTFILE: 表示程序需要读入的文件名
-o OUTPUTFILE: 表示输出文件名,作业提交后的输出到标准输出信息将会保存在这个文件中。
COMMAND: 需要执行的命令
(4) 命令行提交openmpi并行计算作业
openmpi并行需要加上“-a openmpi mpirun.lsf”:
2. LSF脚本提交作业
注意:请咨询系统管理员如何请求计算资源,比如节点、内存与核心数。
(1) 脚本文件的准备
假设每个节点32核,使用8个节点共256核多线程计算作业,脚本文件为foo.lsf,希望提交后的作业名称为HELLO_LSF,要提交一个foo.sh的计算作业。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | #---------这里开始---------- #作业名称 #BSUB -J HELLO_LSF #标准输出 #BSUB -o job.out #错误信息输出 #BSUB -e job.err #请求计算的核心数 #BSUB -n 256 #指定每个节点的核心数为32,也就是说上面的256核从8个32核节点来得 #BSUB -R "span[ptile=32]" #计算命令行,将下面的内容修改为gaussian计算的命令即可 cd /home/gkxiao/demo ./foo.sh #---------这里结束---------- |
假设每个节点32核,使用1个节点共32核多线程计算作业,脚本文件为foo.lsf,希望提交后的作业名称为HELLO_LSF,要提交一个foo.sh的计算作业。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | #---------这里开始---------- ###作业名称 #BSUB -J HELLO_LSF ###标准输出 #BSUB -o job.out ###错误信息输出 #BSUB -e job.err ###请求计算的核心数 #BSUB -n 32 ###指定每个节点的核心数为32,也就是说上面的256核从8个32核节点来得 #BSUB -R "span[hosts=1]" ###计算命令行,将下面的内容修改为gaussian计算的命令即可 cd /home/gkxiao/demo ./foo.sh #---------这里结束---------- |
(2) 提交作业
七. SLURM作业管理系统(同时适用于天河二号)
交互式提交作业
假设作业为Gaussian自带算例的test0397.com,则提交一个单节点、24核心、96GB内存SMP并行的计算作业:
1 | srun -N 1 -n 1 -c 24 g16 -m=96GB -p=24 test0397.com |
等同于:
1 | srun -N 1 -n 24 g16 -m=96GB -p=24 test0397.com |
作业提交后会返回作业号,比如1758829。
有的队列对最长运行时间有约束,则需要指定作业的队列与最长运行时间,此时可以用-p与-t两个选项来配置队列与时长。
1 | srun -p paratera -N 1 -n 1 -c 24 -t 72000 g16 -m=96GB -p=24 test0397.com |
或者:
1 | srun -p paratera -N 1 -n 24 -t 72000 g16 -m=96GB -p=24 test0397.com |
其中-p paratera表示作业提交到paratera队列;-t的单位为分钟,表示运算最长时间为72000分钟(1200小时,50天)。
脚本提交作业
假设作业为Gaussian自带算例的test0397.com,则提交一个单节点、24核心并行计算的脚本,脚本(test0397.sh)内容如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | #!/bin/bash ###set the number of nodes #SBATCH --nodes=1 ### set max wallclock time #SBATCH --time=5000:00:00 ### set name of job #SBATCH --job-name=M01_2 ### mail alert at start, end and abortion of execution #SBATCH --mail-type=ALL ### send mail to this address #SBATCH --mail-user=calc@molcalx.com ### run the application cd $SLURM_SUBMIT_DIR scrdir=`mktemp -d XXXX` export GAUSS_SCRDIR=$SLURM_SUBMIT_DIR/$scrdir g16 -p=24 -m=48GB test0397.com rm -fr $scrdir |
提交作业
1 | sbatch test0793.sh |
这个脚本可以简化为(test0793.sh):
1 2 | #!/bin/sh g16 -p=24 -m=48GB test0793.com |
对应提交作业的方式为:
1 | sbatch -J test0793 -p paratera -N 1 -n 1 -c 24 -t 72000 test0793.sh |
或者:
1 | sbatch -J test0793 -p paratera -N 1 -n 24 -t 72000 test0793.sh |
显示作业详细信息
1 | scontrol show job 1758829 |
取消作业
取消作业号为1758829的作业:
1 | scancel 1758829 |
作业依赖
如果nmr.sh作业需要在另一个作业(比如jobid为2101576)成功后再执行计算,则可以使用作业依赖:
1 | yhbatch -p paratera -N 1 -n 24 -t 72000 --dependency=afterok:2101576 nmr.sh |
八. 跨节点并行
PBS作业管理系统跨节点计算
以PBS作业管理系统为例说明跨节点并行的使用。
1. 软件的安装与普通的Gaussian 16一样
假设Gaussian 16计算环境如下:
1 2 3 | export $g16root=~ export $GAUSS_SCRDIR=~/tmp source $g16root/bsd/g16.profile |
2. 跨节点并行需要额外设置三个环境变量
- (1)节点间通讯协议,通过$GAUSS_LEFLAGS来接收并传给Gaussian
- (2)用环境变量$GAUSS_WDEF接收计算节点,并传递给Gaussian
- (3)用环境变量$GAUSS_PDEF来设定每个节点SMP并行的核心数
比如每个节点12个核心计算:
export GAUSS_PDEF=”12″
3. PBS脚本示例
设定G16环境变量(g16_env.sh)
1 2 3 4 5 6
#---g16_env.sh------- export g16root=/home/test2 export GAUSS_SCRDIR=$g09root/tmp export GAUSS_LFLAGS="-vv -opt 'Tsnet.Node.lindarsharg: ssh'" . $g16root/g16/bsd/g16.profile #---g16_env.sh-------
假设要在2个节点、每个节点12个核心进行并行计算,则PBS脚本如下:
1 2 3 4 5 6 7 8 9 10 11 12 13
#PBS -o /home/zdtest/demo/test0397_pbs.log #PBS -e /home/zdtest/demo/test0397_pbs.err #PBS -l nodes=2:ppn=12 source /home/zdtest/g16_env.sh export GAUSS_WDEF="$(cat $PBS_NODEFILE|sort|uniq|awk '{ printf("%s,",$1) }' | sed 's/,$//')" export GAUSS_PDEF="12" # working directory cd /home/zdtest/demo #run g16 job $g16root/g16/g16 foo.com
SLURM作业管理系统跨节点计算
以SLURM作业管理系统为例说明如何用2个节点共80核心进行跨节点计算。
SLURM脚本test.slurm如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
#!/bin/bash #SBATCH -J gauss16 #SBATCH -p comput #SBATCH -N 2 #SBATCH -n 80 #SBATCH -o out.%j #SBATCH -e err.%j cd $SLURM_SUBMIT_DIR JOB_HOSTLIST=`scontrol show hostnames $SLURM_JOB_NODELIST|sort|uniq|awk '{ printf("%s,",$1) }' | sed 's/,$//'` TASKS_PRENODE=$(($SLURM_NPROCS/$SLURM_JOB_NUM_NODES)) source ~/g16_env.sh export GAUSS_LFLAGS="-vv -opt ‘Tsnet.Node.lindarsharg:ssh’" export GAUSS_WDEF="$JOB_HOSTLIST" export GAUSS_PDEF="$TASKS_PRENODE" export GAUSS_MDEF="128GB" #Run the Gaussian JOB $g16root/g16/g16 uff.com
预先准备的计算文件下载:uff.com
提交作业:
1
sbatch test.slurm
九. GPU的使用
1. 查询GPU设备的基本信息
首先查询一个计算节点有多少张GPU,设备号等基本信息,用nvidia-smi命令即可:
1
nvidia-smi假设你的系统管理员不允许直接到节点查询,可以用作业管理系统的交互式模式查询。查询gpu1队列的节点gpu6的GPU设备,SLURM作业管理系统的命令如下:
1
srun -p gpu1 -w gpu6 nvidia-smi
结果返回:
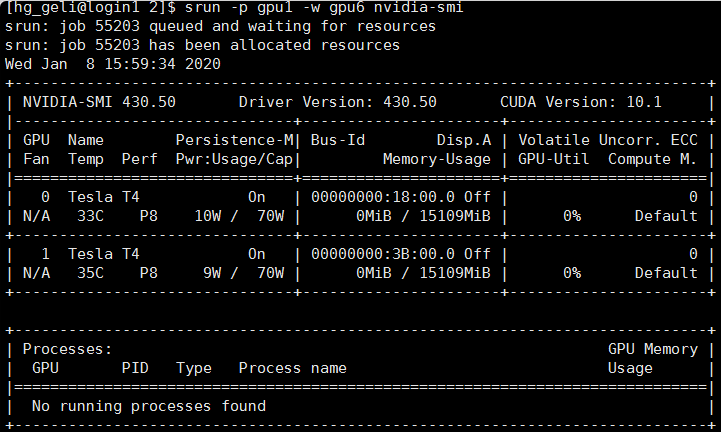
由此可见,在节点gpu6上有两张NVIDIA TESLA T4显卡,设备号(device id)分别为0,1。
以SLURM作业管理系统为例,假设每个节点有2个GPU,这个例子里有两个Tesla T4 GPU,我们演示一下如何用GPU与CPU混合计算。同时,目前这两张显卡处于闲置状态。
2. 查询GPU的控制CPU号
在GPU计算时需要给每个GPU设备安排一个控制CPU。不是每个CPU都可以作为GPU的控制CPU使用。以NVIDIA的CUDA为例,用nvidia-smi命令可以查询这种控制关系:
1
nvidia-smi topo -m
结果如下:
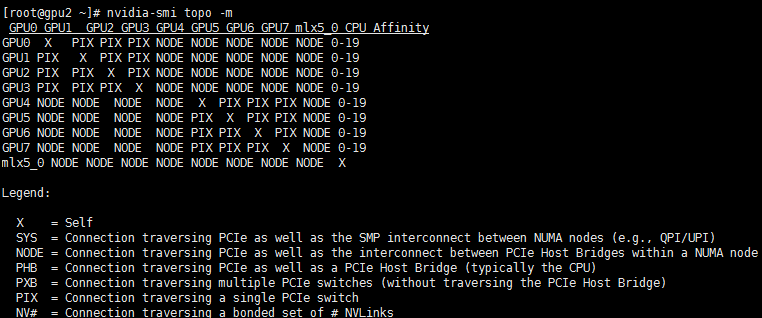
重点关注第1列与最后一列(CPU affinity):第1列给出GPU设备号,CPU Affinity列给出对应的控制CPU。由第1步知道,这个节点有2张GPU,分别是第一列的GPU0与GPU1,他们的控制CPU分别为0-19中的任意两个。
3. 准备Gaussian输入文件
典型的输入如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
%CPU=0-19 %GPUCPU=0,1=0,1 #p opt freq apfd/6-311+g(2d,p) Gaussian Test Job 397: Valinomycin 0,1 O,-1.3754834437,-2.5956821046,3.7664927822 O,-0.3728418073,-0.530460483,3.8840401686 O,2.3301890394,0.5231526187,1.7996834334 O,0.2842272248,2.5136416005,-0.2483875054 .... .... !以上省略的分子描述部分
第1行%CPU=0-19为CPU列表,该列表(从0到19)指示Gaussian程序用第0-19号CPU(共20个CPU)进行计算。第2行%GPUCPU=0,1=0,1,其中第一个等号给出GPU列表,其数值是GPU的设备号,指示Gaussian程序用第0,1号GPU进行计算,这个ID号要与nvidia-smi命令查询出来的一致;其中第二个等号后面为控制CPU列表,其数值对应控制CPU的ID号,而且必须是出现在第1行%CPU的列表里。
4. 提交作业
可以直接提交,也可以写个slurm脚本提交作业:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
#!/bin/bash #SBATCH -J gauss16 #SBATCH -p gpu1 #SBATCH -N 1 #SBATCH -n 20 #SBATCH --gres=gpu:2 #SBATCH -w gpu6 #SBATCH -o out.%j #SBATCH -e err.%j cd $SLURM_SUBMIT_DIR source ~/g16_env.sh JOB_HOSTLIST=`scontrol show hostnames $SLURM_JOB_NODELIST|sort|uniq|awk '{ printf("%s,",$1) }' | sed 's/,$//'` TASKS_PRENODE=$(($SLURM_NPROCS/$SLURM_JOB_NUM_NODES)) export GAUSS_LFLAGS="-vv -opt ‘Tsnet.Node.lindarsharg:ssh’" export GAUSS_WDEF="$JOB_HOSTLIST" #Run the Gaussian JOB $g16root/g16/g16 -m="128GB" test0397.com
再去用nvidia-smi查询节点的GPU状态,会发现两个GPU都被占用了。在Gaussian的输出文件里可以看到相关的如下提示:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52
****************************************** Gaussian 16: ES64L-G16RevC.01 3-Jul-2019 11-Jan-2020 ****************************************** %mem=128GB %cpu=0-19 SetSPE: set environment variable "MP_BIND" = "yes" SetSPE: set environment variable "MP_BLIST" = "0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19" Will use up to 20 processors via shared memory. %GPUCPU=0,1=0,2 Will use 2 GPUs: Thread CPU GPU 0 0 0 1 2 1 2 1 -1 3 3 -1 4 4 -1 5 5 -1 6 6 -1 7 7 -1 8 8 -1 9 9 -1 10 10 -1 11 11 -1 12 12 -1 13 13 -1 14 14 -1 15 15 -1 16 16 -1 17 17 -1 18 18 -1 19 19 -1 SetGPE: set environment variable "MP_BIND" = "yes" SetGPE: set environment variable "MP_BLIST" = "0,2,1,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19" SetLPE: input flags="-vv -opt ‘Tsnet.Node.lindarsharg:ssh’ " SetLPE: new flags="-vv -opt ‘Tsnet.Node.lindarsharg:ssh’ -opt 'Tsnet.Node.lindarsharg: ssh' -nodelist 'gpu4' -env GAUSS_MDEF=17179869184 -env GAUSS_EXEDIR="/public/home/hg_geli/g16/bsd:/public/home/hg_geli/g16" -env "MP_BIND=yes" -env "MP_BLIST=0,2,1,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19" -env GAUSS_ADEF="0,2,1,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19" -env GAUSS_BDEF="0,1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1,-1"" Will use up to 1 processors via Linda. Properties for Device 0 Minimum vector length: 32 Maximum vector length: 1024 Number of SMPs: 40 Maximum number of threads per SMP: 1024 Number of ASync Engines: 3 Memory: 1161412608 Properties for Device 1 Minimum vector length: 32 Maximum vector length: 1024 Number of SMPs: 40 Maximum number of threads per SMP: 1024 Number of ASync Engines: 3 Memory: 872792064 785512857 words of memory will be used on each GPU.
十. 上门安装技术服务
如果您需要上门安装、集群技术服务,我们会为您推荐第三方上门服务(包含集群维护服务)。
十一. 问题反馈
请联系我们:020-38261356 info@molcalx.com
export GAUSS_LFLAGS=”-vv -opt ‘Tsnet.Node.lindarsharg: ssh'”
PBS脚本从管理系统获取计算资源,并传递给Gaussian,从PBS的管理器获取节点
export GAUSS_WDEF=”$(cat $PBS_NODEFILE|sort|uniq|awk ‘{ printf(“%s,”,$1) }’ | sed ‘s/,$//’)”


