
摘要:除了在硫与氮/氧孤对电子之间发生有利的相互作用之外,在硫原子与氧或氮原子的孤对电子之间还可能发生对结合不利的排斥相互作用,这使得最终的净相互作用对结合有利是否难以判断。本文以一对活性差异5 kcal/mol的CLK抑制剂分子对4、7为例,其中7可视为用硫-氧/氮孤对电子相互作用代替化合物4经典氢键相互作用的设计,但这导致7的活性丧失。然而,在本文中,基于几何条件的相互作用分析以及分子对接打分未能区分4、7的活性差异。而基于Cresset XED力场的蛋白相互作用势与配体场不仅可以可视化分析经典的氢键、非经典的C-H氢键以及硫-氧/氮孤对电子相互作用,还可以通过配体场在关键位点的场值来解释4与7的之间活性差异。基于SQM的相互作用能可以进一步给出了定量分析结果,并与实验观察到的活性差异一致。总地来说,本文的研究表明,Cresset XED场技术与SQM ΔEinter为研究硫-氧/氮孤对电子相互作用提供了一个可靠的工具。
肖高铿/2024-09-08
前言
Leucettinib-21(化合物4,图1-左)是Lindberg等人1从670多个Leucettinibs衍生物中筛选出来的一种治疗唐氏综合症(Down syndrome, DS)和阿尔茨海默病(Alzheimer’s disease, AD)的临床在研究药物,其靶标为双特异性、酪氨酸磷酸化调节激酶1A(DYRK1A)。
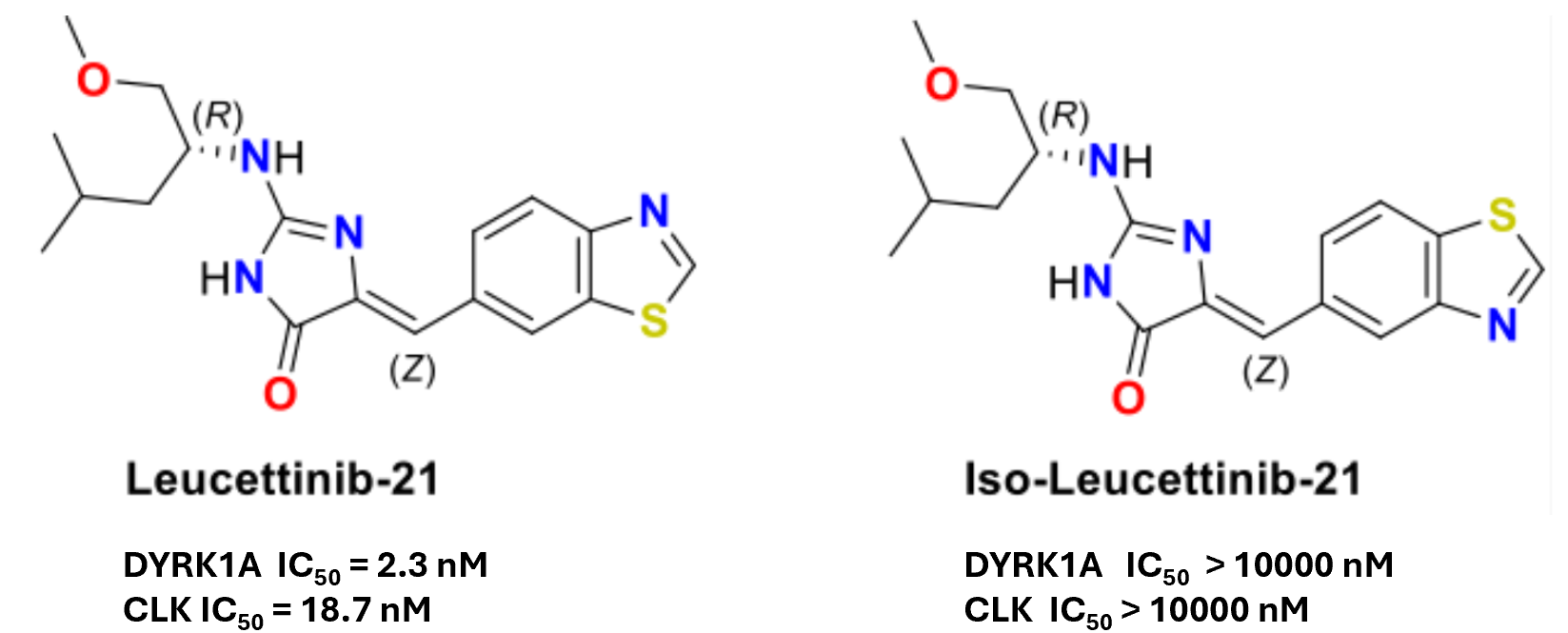
图1. Leucettinib-21及其非活性异构体的化学结构式。左:化合物4;右:化合物7
单晶衍射数据表明4存在三种不同的异构体形式1,如图2所示,这些异构体是由于分子中氨基咪唑酮片段氨基氢原子的重新定位而产生的。对三个异构体模型进行精修,与初始模型相比,R因子显著下降,这三种异构体在晶体结构中大致以相等的比例存在,比例约为0.29/0.41/0.30。在所有三种异构体中,尾部的(R)手性都得到了保留。
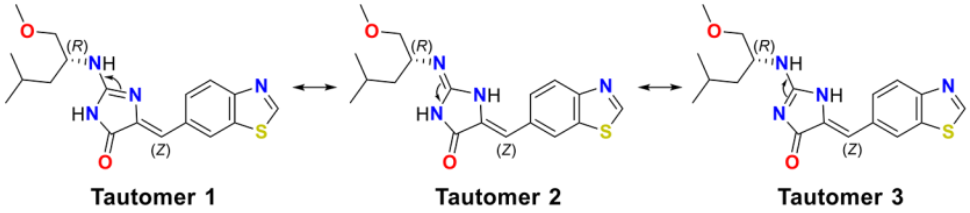
图2. 化合物4的三种互变异构体
DYRK1A与CLK具有高度相似的结合口袋,因此化合物4除了是DYRK1A的抑制剂(IC50=2.4nM)之外,还是CLK的抑制剂(IC50=18.7nM)。比较有意思的是,其同分异构体Iso-Leucettinin-21(化合物7,图1-右)与4仅在噻唑环上的N/S原子位置差异,但却几乎丧失了对DYRK1A(IC50\( \gt \)10000nM)与CLK(IC50\( \gt \)10000nM)的活性,并在研究中作为阴性对照药。
从化学结构上看,化合物4与7差异仅在于噻唑环上的氮/氧原子位置、但这导致了对CLK的IC50存在530多倍的活性差异,鉴于作者解释了其中4与CLK的共晶结构PDB 8P08,这构成了一个可从结构生物学角度研究活性悬崖的分子对,这对了解硫-孤对电子相互作用与经典氢键相互作用的差异提供了非常难得的素材。本文的目的是,尝试利用可视化分析、半经验量子力学计算与基于结构的方法来解释化合物4与7对CLK抑制活性差异的原因。
结果
化合物4、7与CLK的相互作用分析
化合物4与CLK的结合模式根据共晶结构PDB 8P08而来,化合物7与CLK的结合模式是用FlareTM V82在CLK的结合口袋里对7进行编辑、力场几何优化而来。化合物4、7与CLK相互作用的3D与2D图分别如图3、4所示。
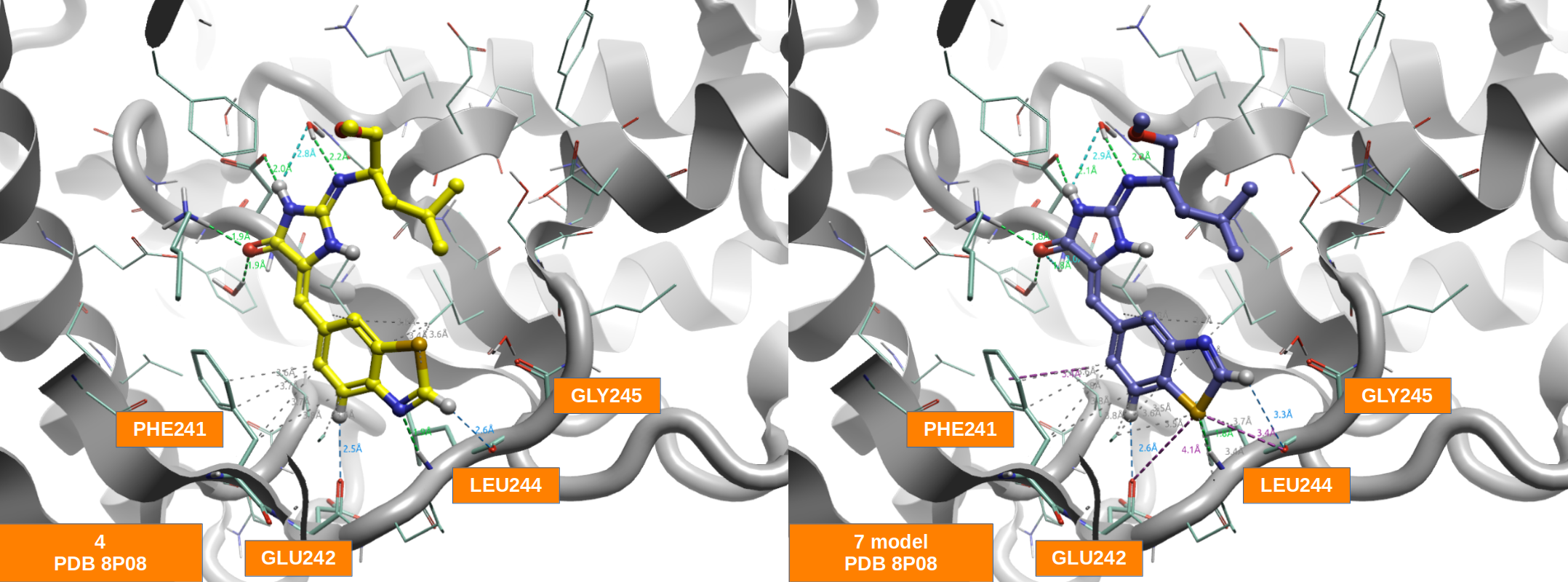
图3. 化合物4、7与CLK的结合模式。黄色:化合物4;青灰色:化合物7;灰色飘带:CLK。
其中,咪唑酮片段的质子化状态采用了单晶衍射实验发现的丰度最高的异构体形式(图2-Tautomer 2)。我们重点关注苯并噻唑片段,它指向铰链结合区。化合物4的噻唑氮原子作为氢键受体与LEU244的蛋白骨架NH形成氢键相互作用,除此之外,没有看到任何经典的、强极性相互作用。化合物7的苯并噻唑的硫作为氢键受体与LEU244的蛋白骨架NH形成氢键相互作用,虽然也没有看到任何其它经典的、强极性相互作用,但是噻唑的硫原子与GLU242、LEU244的羰基氧发生了硫-氧孤对电子(lone pair,lp)相互作用,这让人禁不住期待4、7具有相似活性的可能。
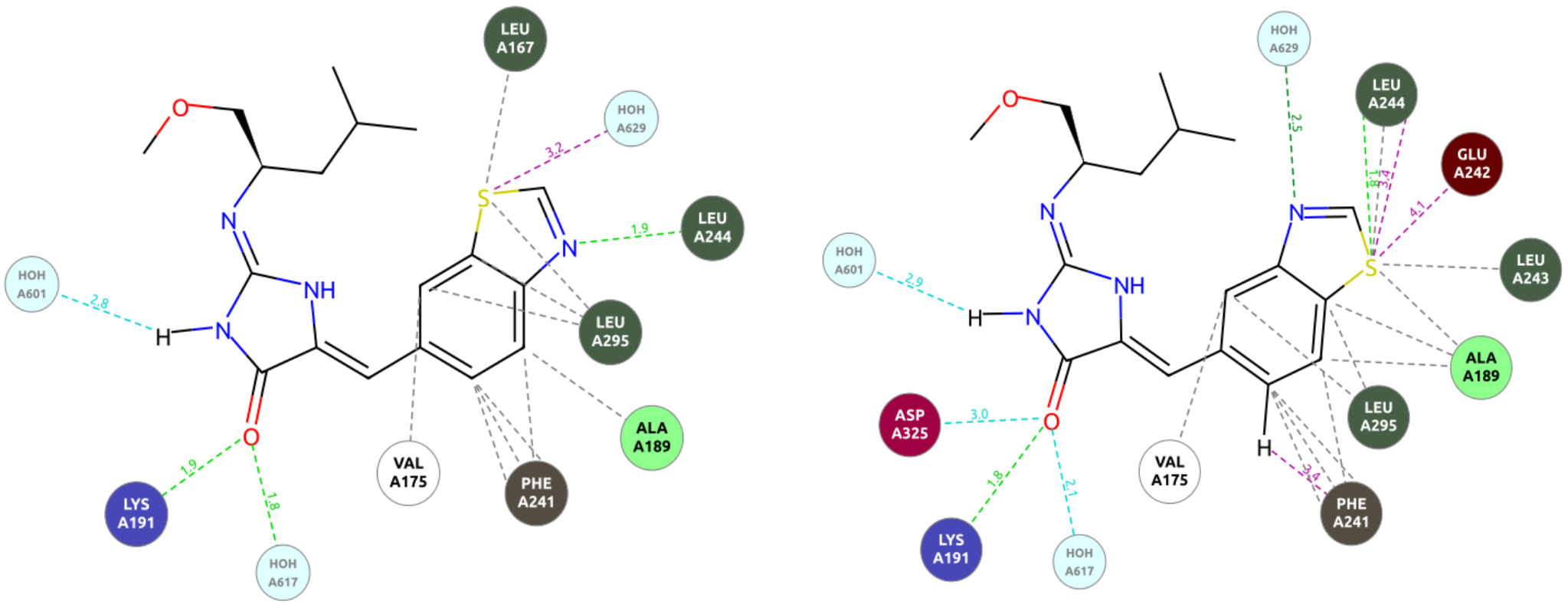
图4. 化合物4、7与CLK的结合模式2D图。
现有的研究表明3-5,硫-孤对电子相互作用的几何要求包括S···A Distance = 2.7-4.2 Å以及∠X-S···A \(\gt 140˚\)。如图5所示,Flare的Contact Analysis捕捉到在化合物7的噻唑与铰链区之间有两个满足几何要求的硫-孤对电子相互作用。其中之一是在苯并噻唑硫与Glu242羰基氧之间,S···O d = 4.3 Å以及∠C-S···O \(= 147˚\);另一个是在苯并噻唑硫与Leu242的羰基氧之间,S···O d = 3.2 Å以及∠C-S···O \(= 163˚\),氧原子几乎位于苯并噻唑C-S的延长线上。前者看起来刚刚满足几何要求,而后者则要相当完美地符合要求,这让人怀疑两者在相互作用强度上是否有显著区别,但没法从几何模式中直接判断。
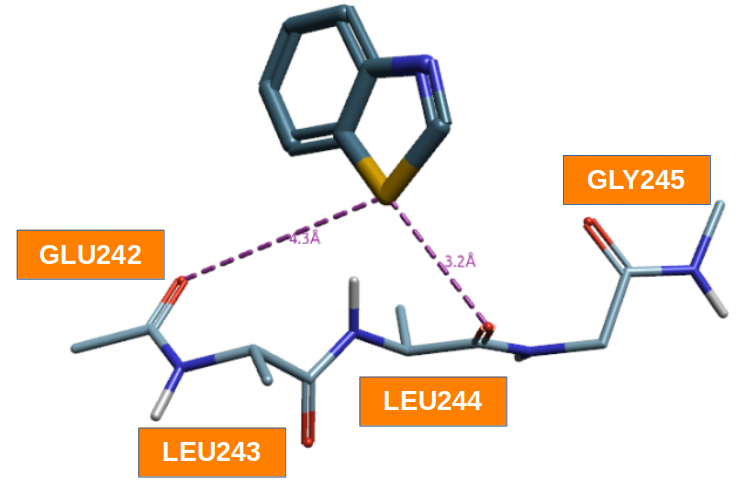
图5. 化合物7的苯并噻唑与铰链区之间的硫/孤对电子相互作用示意图
Hudson等人6用自然键轨道分析方法归属了孤对电子(lone pair,lp)之间不利的相互作用以及硫-孤对电子有利相互作用的成分与来源。研究表明,除了在硫与氮/氧孤对电子之间存在有利的相互作用之外,在硫原子的孤对电子与氧或氮原子的孤对电子之间还具有不利排斥的相互作用。因此,化合物7能否与4一样具有相当的活性,还取决于硫与氮/氧孤对电子之间的有利相互作用能否对不利的孤对电子排斥作用提供补偿并获得净有利的相互作用。显然,4、7与CLK铰链区在相互作用上的差异是它们活性差异的原因,这需要借助合适的工具用来权衡这些有利的与不利的相互作用,并用来解释活性差异。
蛋白相互作用势(PIP)与配体场可视化定性分析
Cresset XED力场7,8通过复杂的原子描述来模拟远离原子中心的电荷,这使得能够更详细地描述静电并优异地重现分子间相互作用。
首先用Flare V82计算PDB 8P08″干”结合位点的PIP,为了清晰起见,未展示蛋白结构,结果如图6(上)所示。有三个PIP等值图与配体的苯并噻唑片段重合,分别编号为-ev 1, +ev 2与-ev 3。+ev代表正的PIP,-ev代表负的PIP。图6-下显示了配体4、7的配体场,红色等值图表示正静电势,蓝色表示负静电势。
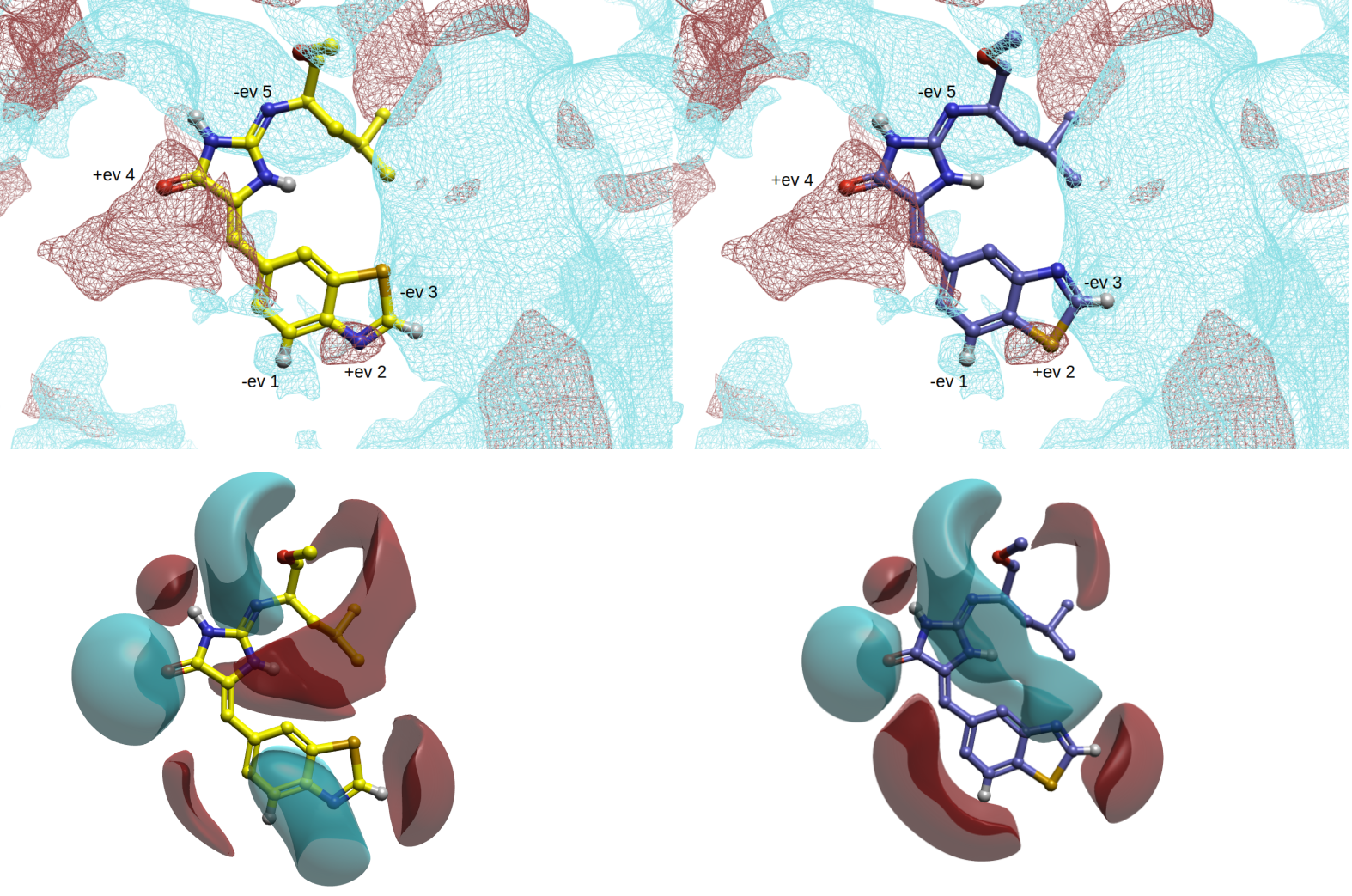
图6. PDB 8P08“干”蛋白的结合位点的PIP(上)与配体场(下)。红色网格:正PIP,level = 4.0;蓝色网格:负PIP,level = -2.0;黄色配体:4;青灰色配体:7;红色实心区块:+ev配体场(threshold = 2);蓝色实心区块:-ev配体场(threshold = 2)。
我们可以发现,配体的苯并噻唑环(图6-下)的静电场与蛋白的PIP是互补的。比如,苯并噻唑的苯环C-H为配体场正静电势区,被编号为负PIP区包埋,蛋白与配体在静电上是互补的;噻唑环上的C-H处有着很强的红色正静电势区,被编号为-ev 3的蓝色负PIP区包埋,因此配体-蛋白从静电上也是互补、相互作用有利的。化合物4、7最大差异在于蛋白PIP的+ev 2处,它与化合物4的噻唑氮原子附近的蓝色负配体场互补;而化合物7在该处为S原子,出现一个空白的孔洞,显而易见地,7的配体场与编号为+ev 2的蛋白PIP的互补性不是很好。实际上,+ev 2的PIP对应于铰链区LEU244的NH氢键供体,这意味着化合物4与7与该氢键供体的相互作用存在显著的差异,提示这是化合物7活性差的主要原因之一。
配体场定量分析
除了上述基于XED力场的配体场定性可视化分析之外,还可以用pyflare计算在铰链区关键原子上的来自配体的静电场来半定量分析化合物4、7差异,结果如图7所示。
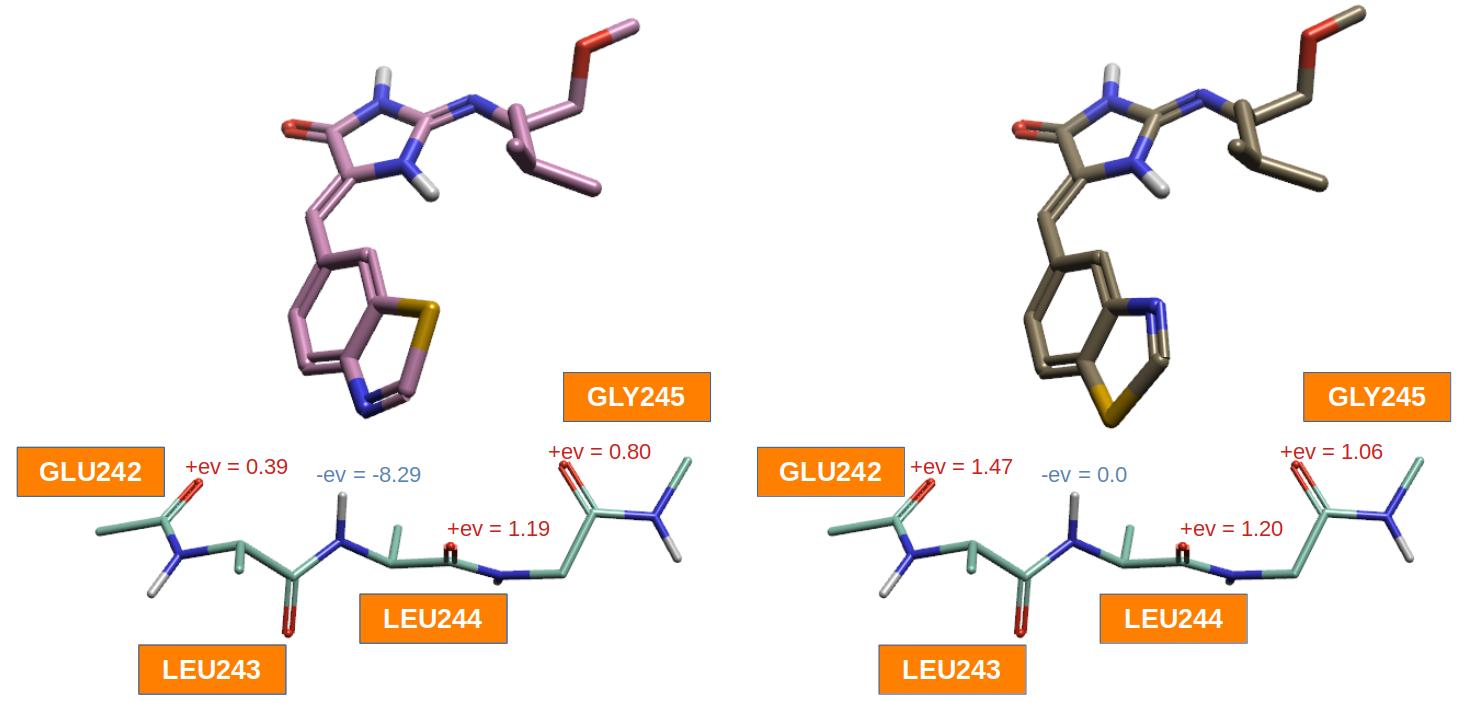
图7. 化合物4、7在铰链区关键氢键受体羰基氧与氢键共体NH原子上的XED静电场值
首先注意到在铰链区LEU244-NH氢原子处,化合物4、7的配体静电场分别为-8.29与0,说明化合物4比7更有利于与LEU244-NH发生有利的相互作用。LEU244-NH氢原子处的配体场值同时也与图6(下)所示的配体场一致:化合物4噻唑氮原子有着浓郁的蓝色-ev区,而7噻唑硫原子在该处呈现空白。静电场值为4、7的相互作用差异提供了直观的“数值”参考。
在铰链区GLU242的羰基氧原子处,化合物4、7的配体静电场分别为+0.39与1.47,这与图6(下)观察到4在苯环C-H附近的红色配体场不如7的大是一致的。一方面,该处的正静电场与苯环的C-H作为氢键供体的作用一致,另一面7比4在此处多了来自噻唑C-S方向有利的硫-孤对电子相互作用。两者的加和使得7比4具有更强的正静电场值。
在铰链区LEU244羰基氧处,化合物4与7具有相似的配体静电场,分别为+1.19与1.20,前者与噻唑C-H氢键供体有关,后者与噻唑S···O孤对电子相互用相关。相似的正配体场值可能意味着两者与LEU244羰基氧的相互作用相当。在铰链区GLY245羰基氧上,化合物4、7的配体静电场分别为+0.80与1.06。如图6所示,LEU244与GLY245的羰基氧均被噻唑C-H方向的配体正静电场覆盖,其中7的红色配体静电场往GLY245羰基氧方向上更加深厚一点,这与GLY245羰基氧上配体场值差异趋势一致。
总的来说,配体场的定量分析可以增强配体可视化分析效果,尤其在解释C-H···O=C氢键相互作用与S···O孤对电子相互作用方面提供了“数值”参考,尤其在比较关键相互作用位置上作用强度差异时很有用。
用SQM进行相互作用分析
化合物4、7铰链结合片段噻唑环的取向不同是这两个化合物与CLK活性差异的原因所在,其中7还涉及复杂的S···O/LP相互作用,这用力场通常难以准确描述5,因此我们决定用半经验量子力学(Semiempirical Quantum Mechanics,SQM)来定量计算4、7与CLK的相互作用能(ΔEinter):
$$
ΔE_{inter} = E_{complex} – E_{hinge} – E_{ligand} \cdots (1)
$$
其中Ecomplex、Ehinge与Eligand分别表示在SQM理论水平计算的抑制剂-铰链区残基、铰链区残基、配体的能量。
其中用来计算相互作用能的铰链区氨基酸残基为GLU242-LEU243-LEU244-GLY245,用Flare从PDB 8P08提取出来,并进行结构准备,封端。铰链区进一步在BP86-D3/DEF2-SVP理论水平对氢进行几何优化之后用于SQM单点能计算与相互作用能计算。这里,没有用整个蛋白结构和更小一点的结合位点来计算与配体的相互作用,一方面是因为出于计算速度考虑,另一方面我们仅对涉及与苯并噻唑发生相互作用的Hinge残基感兴趣以确保计算反应差异。
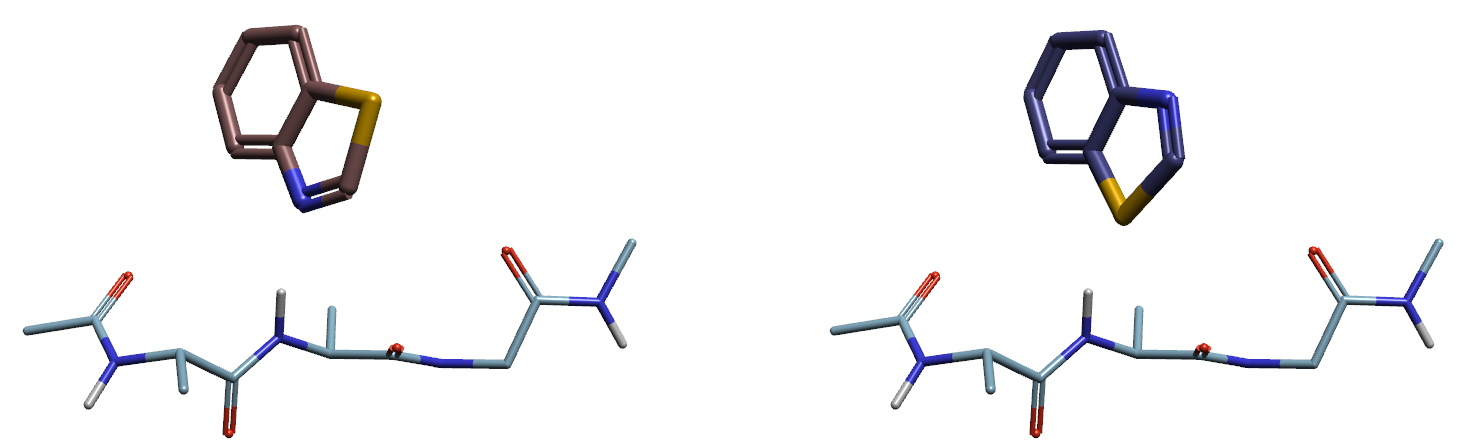
图8. SQM ΔEinter计算的模型分子。棍棒模型-左:化合物4;棍棒模型-右:化合物7;细线:铰链区残基。
其中化合物4、7用其模型分子苯并噻唑代替(图8),在BP86-D3/DEF2-SVP理论水平进行无约束的优化后再叠合PDB 8P08的共晶配体上,然后再SQM理论水平进行单点能计算铰链区-配体的复合物能量与配体的能量。两种SQM程序用来计算ΔEinter,一种是用MOPAC9在PM7-MOZYME理论水平计算,一种是用xTB在GFN2-xTB理论水平计算10-12,结果如表1所示。
表1. 两种SQM方法计算4、7与CLK的相互作用能
| Item | MOZYME-PM7(kcal/mol) | GFN2-xTB(kcal/mol) | CLK Exp. ΔGBinding (kcal/mol) |
|---|---|---|---|
| Ehinge | -198.10 | -38751.80 | |
| E4 | 51.57 | -15870.85 | |
| E4-hinge | -158.09 | -53633.17 | |
| 4 ΔEinter | -11.56 | -10.52 | -11.78 |
| E7 | 51.57 | -14870.85 | |
| E7-hinge | -153.40 | -53615.21 | |
| 7 ΔEinter | -6.87 | 7.44 | \(\gt-6.81\) |
\(ΔG_{binding} = RTlnIC_{50}\)
从表1可以看出,化合物4与7的结合自由能(根据IC50计算)差值与MOPAC、xTB计算的SQM ΔEinter差值非常一致,这有力地支持了前面关于化合物4、7的活性差异取决于苯并噻唑与铰链区相互作用差异的假设。
用分子对接打分函数进行相互作用分析
GNINA13,14是基于流行分子对接软件AutoDock Vina15的深度学习打分版本。用GNINA实现的Vina score与CNN affinity在score only模式下预测化合物4、7与CLK的结合自由能和结合亲合力,结果如表2所示。
表2. 化合物4与7的分子对接打分结果
| Items | 4 | 7 |
|---|---|---|
| gauss 1 | 89.27723 | 85.56207 |
| gauss 2 | 1491.20154 | 1490.39343 |
| repulsion | 2.90202 | 4.05842 |
| hydrophobic | 44.70626 | 44.96795 |
| H-Bond | 2.79982 | 1.79055 |
| Vina score (kcal/mol) | -8.62 | -7.36 |
| CNNaffinity(pKd) | 7.83 | 7.59 |
| CNNscore | 0.96 | 0.90 |
| CLK Exp. IC50 (nM/pKd) | 2.3/8.6 | \(\gt10000\)/5.0 |
| CLK Exp. ΔGbinding(kcal/mol) | -11.78 | \(\gt-6.81\) |
注:AutoDock Vina的打分项为加权之前的值;其中\(pK_d=-{log_{10}{IC_{50}}}\),\(ΔG_{binding} = RTlnIC_{50}\)。
GNINA的CNN score评估了结合模式为native pose的概率,4与7的CNN score均在0.9以上,说明起始用于打分的pose具有很高的可信度。化合物4比7具有略高的gauss1与氢键相互作用项,而且还有具有更小的排斥项,这导致化合物4比7的Vina score(结合自由能预测值)低了1.4kcal/mol,然而这个差值不足以说明在实验上观察到的5kcal/mol差异。虽然CNN affinity预测4的结合亲合力(pKd=7.83)与实验值(pKd=8.6)相当一致,但对没有实验活性的7也给出相似的预测值(pKd=7.59),这说深度学习打分函数CNN affinity也未能区分4与7之间高达3.6个数量级的亲和力差异。
用基于XED的配体场与PIP、以及SQM ΔEinter来增强硫-孤对电子相互作用分析
通过几何条件的满足情况来发现蛋白-配体相互作用中有利的硫-氮/氧孤对电子相互作用是常规的方法。然而,在硫的孤对电子与氮/氧的孤对电子之间也可能同时发生不利的相互作用,只有当有利的相互作用大于不利的相互作用时,硫-氮/氧孤对电子相互作用才能为结合带来净有利的效果。这种不利的孤对电子间相互作于通常不容易分析,也容易被忽略。在本文中,4与7苯并噻唑取向的不同,导致4与铰链区发送经典氢键相互作用,而7虽然少了该氢键相互作用、但是多了两个硫-氧孤对电子相互作用。常规的方法难以解释4与7之间高达5 kcal/mol的结合自由能差异。基于XED的配体场与蛋白相互作用场(PIP)可视化分析不仅可以洞察经典的氢键相互作用,而且还可以洞察非经典的C-H氢键以及硫-氧/氮孤对电子相互作用,尤其当与基于配场的定量分析组合使用时可以很好的解释4与7的之间活性差异。基于SQM的相互作用能(ΔEinter)分析进一步给出了定量分析结果,并与实验观察到的5kcal/mol活性差异一致。而流行的分子对接打分函数Vina score与深度学习打分CNN affinty均未能将化合物4、7的活性区分开。总的来说,基于XED的场技术与SQM ΔEinter可以增强硫-孤对电子相互作用分析,更好地解释实验观察到的结果。
方法
蛋白与配体的结构准备
将共晶结构从PDB下载到FlareTM V82中,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,任何截短的蛋白质链被封端作为蛋白质准备的一部分。
用MOPA在PM7-MOZYME理论水平计算复合物结构、Hinge与配体的生成焓
以化合物7的复合物为例,在PM7-MOZYME理论水平计算生成焓的关键次如下第1行:
1 2 3 4 5 6 7 8 9 10 11 | PM7 CHARGE=0 MOZYME 7-complex C -17.91160 1 5.81170 1 17.03930 1 C -18.83110 1 5.09670 1 17.82920 1 C -19.49460 1 5.70560 1 18.89060 1 C -19.21990 1 7.05250 1 19.14990 1 C -18.87050 1 9.39190 1 19.75340 1 C -18.29260 1 7.78720 1 18.35930 1 ... !此处省略了原子坐标 |
计算完毕,在输出文件里找‘FINAL HEAT OF FORMATION’行,结果看起来如下:
1 | FINAL HEAT OF FORMATION = -153.40259 KCAL/MOL = -641.83644 KJ/MOL |
其中-153.40259就是该体系的生成焓,用来计算相互作用能。
用xTB在GNF2-xTB理论水平计算复合物结构、Hinge与配体的电子结构能量
将在DFT理论水平优化好结构保存为xyz格式文件,直接用xTB在默认参数下单点计算。以化合物7复合物结构的单点能计算为例:
1 | xtb 7_complex.xyz --chrg 0 > 7_complex_sp.out |
计算完毕,在输出文件里找‘TOTAL ENERGY’行,结果看起来如下:
1 2 3 4 5 | -------------------------------------------------
| TOTAL ENERGY -85.442569915855 Eh |
| GRADIENT NORM 0.140671778122 Eh/α |
| HOMO-LUMO GAP 3.010831241729 eV |
------------------------------------------------- |
在xTB里,电子结构能量的单位Hartree,因此需要转化为kcal/mol。
Vina与GNINA CNN打分
AutoDock Vina打分13与GNINA的深度学习CNN打分采用GNINA在score only模式下计算14,15。
用pyflare计算配体场
用鼠标选择一个感兴趣的点,比如LEU244-NH上氢,计算化合物4、7在该点的XED静电场,可以用下面pyflare代码实现。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | # Documentation can be found at Python > Documentation. # This default python script can be edited at: # '/home/gkxiao/.com.cresset-bmd/Flare/python/default-scripts/InterpreterDefaultScript.py' from cresset import flare p = flare.main_window().project # for positive field type #FP_TYPE = flare.FieldType.Pos # for negative field type FP_TYPE = flare.FieldType.Neg atm1 = flare.main_window().picked_atoms[0] ligs = p.ligands for lig in ligs: fieldval = lig.field_value(FP_TYPE,atm1.pos) lig.properties['FieldVal'].value = fieldval |
结论
用与蛋白发生硫-氧/氮孤对电子相互作用的片段来代替氢键相互作用有可能在保留活性的前提下改善化合物的DMPK性质。然而,除了在硫与氮/氧孤对电子之间发生有利的相互作用之外,在硫原子与氧或氮原子的孤对电子之间还可能发生对结合不利的排斥相互作用,这使得最终的净相互作用对结合是否有利难以判断。
化合物4、7是一对仅在噻唑环的氮/原位置存在差异的同分异构体,可以将化合物7视为用硫-氧/氮孤对电子相互作用代替化合物4经典氢键相互作用的设计,但这使得7丧失了对CLK的抑制活性,4与7的结合自由能差异高达5 kcal/mol。然而,在本文中,基于几何条件的相互作用分析以及流行的分子对接打分Vina Score与GNINA CNN affinity未能区分4、7的活性差异。而基于Cresset XED力场的蛋白相互作用势(PIP)与配体场不仅可以可视化分析经典的氢键、非经典的C-H氢键以及硫-氧/氮孤对电子相互作用,还可以通过配体场在关键位点的场值来解释4与7的之间活性差异。基于SQM的相互作用能(SQM ΔEinter)分析进一步给出了定量分析结果,并与实验观察到的5kcal/mol活性差异一致。
总地来说,本文的研究表明,Cresset XED场技术与SQM ΔEinter为研究硫-氧/氮孤对电子相互作用提供了一个可靠、便利的工具。
文献
- Lindberg, M. F. et al. Chemical, Biochemical, Cellular, and Physiological Characterization of Leucettinib-21, a Down Syndrome and Alzheimer’s Disease Drug Candidate. Journal of Medicinal Chemistry 66, 15648–15670 (2023).
- Flare™, version 8.0.1, Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/flare/; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation J. Chem. Inf. Model. 2006, 46 (2), 665-676; Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes J. Med. Chem. 2019, 62, 6, 3036-3050; Maximilian Kuhn, Stuart Firth-Clark, Paolo Tosco, Antonia S. J. S. Mey, Mark Mackey and Julien Michel Assessment of Binding Affinity via Alchemical Free-Energy Calculations J. Chem. Inf. Model. 2020, 60, 6, 3120–3130
- R. Beno, B. et al. (2015) “A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design,” Journal of Medicinal Chemistry, 58(11), pp. 4383–4438. Available at: https://doi.org/10.1021/jm501853m.
- Zhang, X. et al. (2015) “Intermolecular Sulfur⋯Oxygen Interactions: Theoretical and Statistical Investigations,” Journal of Chemical Information and Modeling, 55(10). Available at: https://doi.org/10.1021/acs.jcim.5b00177.
- Koebel, M.R. et al. (2016) “S⋯O and S⋯N Sulfur Bonding Interactions in Protein–Ligand Complexes: Empirical Considerations and Scoring Function,” Journal of Chemical Information and Modeling, 56(12), pp. 2298–2309. Available at: https://doi.org/10.1021/acs.jcim.6b00236.
- Hudson, B.M., Nguyen, E. and Tantillo, D.J. (2016) “The influence of intramolecular sulfur–lone pair interactions on small-molecule drug design and receptor binding,” Organic & Biomolecular Chemistry, 14(16), pp. 3975–3980. Available at: https://doi.org/10.1039/C6OB00254D.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. II. Organic Complexes. J. Comput. Aided. Mol. Des. 1996, 10 (5), 417–426. https://doi.org/10.1007/BF00124473.
- Cresset的核心技术. 墨灵格的博客. http://blog.molcalx.com.cn/2019/10/08/cresset-science-xed.html
- Moussa, Jonathan E. and Stewart, James J. P. (2024) MOPAC Version 22.1.1. https://github.com/openmopac/mopac
- Bannwarth, C., Ehlert, S. and Grimme, S. (2019) “GFN2-xTB – An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions,” Journal of Chemical Theory and Computation, 15(3). Available at: https://doi.org/10.1021/acs.jctc.8b01176.
- Bannwarth, C. et al. (2021) “Extended tight-binding quantum chemistry methods,” Wiley Interdisciplinary Reviews: Computational Molecular Science. Blackwell Publishing Inc. Available at: https://doi.org/10.1002/wcms.1493.
- https://github.com/grimme-lab/xtb
- Sunseri, J., & Koes, D. R. Virtual Screening with Gnina 1.0. Molecules, 2021, 26(23), 7369. https://doi.org/10.3390/molecules26237369
- GNINA 1.0, https://github.com/gnina
- Trott, O. and Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 2009, 31(2), 455–461. Available at: https://doi.org/10.1002/jcc.21334.
联系我们、获取试用或商务合作
想要亲自计算或在自己的项目中使用Flare,或者商务合作,请联系我们。


