
摘要:FRED是OpenEye OEDocking软件包的一个分子对接程序,它将化合物数据库对接到一个受体口袋里进行结合模式预测、虚拟筛选等。本教程讲解了如何用FRED进行对接计算,并以ABL激酶及其抑制剂格列卫相互作用模式预测为例讲解了FRED的操作步骤:1)受体结构准备;2)配体结构准备;3) 分子对接;4) 结果分析。
肖高铿/2016-09-15
一.FRED-分子对接简介
FRED是OpenEye的OEDocking对接软件包中的一个子程序。FRED的主要功能是进行分子对接计算:将一个分子或化合物数据库对接到指定蛋白的结合位点,进行虚拟筛选。整个计算过程仅利用到了一个蛋白的结合位点信息与数据库的化合物结构。
分子对接预测配体-受体结合模式(binding mode)的问题其实是预测pose的问题:配体在结合位点里会以何种姿势与受体相互作用呢?FRED采用穷尽搜索算法(见图1),即系统性地枚举配体每个构象在特定分辨率的结合位点里的平动与转动空间。在穷尽搜索的时候,将不可能的pose过滤掉,对保留下来的进行打分。搜索完毕,对打分最高的那些poes进一步进行优化(局部穷尽搜索,比全局穷尽搜索用更高的分辨率),最后打分最高的那个pose与数据库中其它化合物进行比较、排序,完成虚拟筛选。
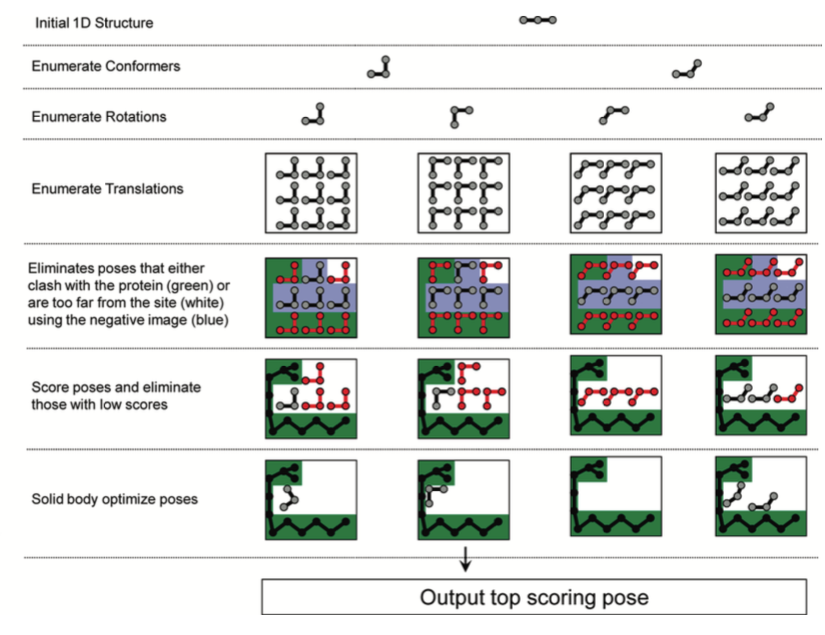
图1. FRED的算法(Mark McGann 2011)
FRED对接的整个过程中蛋白是刚性的,配体的每个构象也是刚性的,配体的柔性是隐式包含在预先的配体构象系综中(对接时输入的是预先构象搜索好的全部低能构象)。因为化合物的构象生成不依赖于对接的结合位点,所以FRED分子对接的虚拟筛选可以极大的节省对接计算时间。在OpenEye的分子对接流程中,OMEGA用于对接前的构象搜索:根据Paul C. D. Hawkins(2010)等人的研究,OMEGA可以高效地搜索构象、并在低能构象中包含有一个生物活性构象(见图2)。
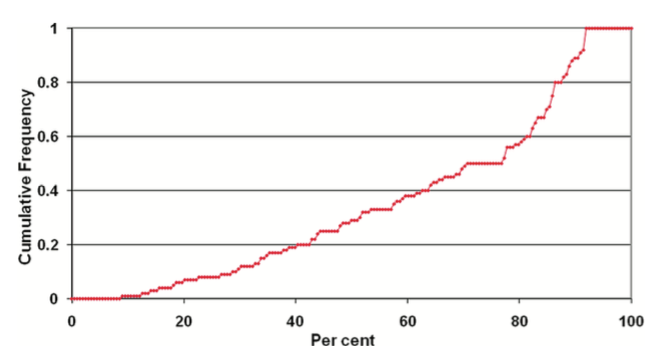
图2.在测试中85%以上测试集的生物活性构象被OMEGA在1.5埃的误差内重现出来
二. 场景假设
- 已有配体-受体复合物3D结构,将一个化合物/数据库对接到复合物共晶配体的结合位点。
- 没有配体-受体复合物结构,仅有受体的3D结构, 根据氨基酸残基列表来定义结合位点,将一个化合物/数据库对接到结合位点。
- 已经分别准备好了受体结构文件(比如:receptor.mol2)与配体文件(ligand.mol2)或box文件(box.pdb),需要将一个化合物/数据库对接到复合物共晶配体的结合位点。
配体-受体的复合物结构文件可以从PDB数据库下载,FRED支持从复合物晶体结构出发准备受体文件进行虚拟筛选。
FRED分子对接仅利用蛋白的3D结构信息,因此,通过氨基酸残基列表与蛋白3D结构,即可准备受体文件进行基于结构的虚拟筛选。
诸如PDB Bind与scPDB等深度注释的蛋白结构数据库已经将复合物结构拆解为单独的受体结构文件与共晶的配体文件,FRED还支持从这样的受体与配体文件出发准备对接用受体文件进行虚拟筛选。
三. 操作步骤
- 受体文件的准备
- 配体结构准备
- 分子对接(虚拟筛选)
- 结果分析
受体文件是一个特殊的OEBinary文件(.oeb,.oeb.gz文件),它包含了靶蛋白的结构以及额外的关于结合口袋位置与特征的信息。OEDocking软件包的每个对接程序(FRED, HYBRID, POSIT)都要用到这个受体文件。FRED分子对接也不例外,受体文件准备方法见表1。
表1. 分子对接受体结构的准备
| 命令 | 应用场合 |
| pdb2receptor | 将配体-受体复合物PDB结构转化为分子对接计算用的受体文件(receptor file);或者指定一个或几个氨基酸残基做为结合位点准备对接计算用的受体文件; |
| receptor_setup | 将一个蛋白结构及其共晶的配体或覆盖结合位点的盒子转化为分子对接用的受体文件。与pdb2receptor不同的是,receptor_setup需要将配体-受体复合物拆分成两个文件:一个是受体蛋白结构文件,另一个是共晶的配体文件或box文件; |
| apopdb2receptor | 将非结合蛋白(apo protein)转化为分子对接用的受体文件(receptor file)。活性位点(active site)由用户通过氨基残基来指定(-site_residue)。氨基酸残基做为活性位点的提示进行口袋/沟槽遗迹结合腔进行搜索计算。 |
| make_receptor | 受体结构准备图形用户界面,不是命令行。 |
(1) pdb2receptor
要求PDB结构必须为受体配体复合物结构,以共晶的配体来指示结合位点。假设PDB仅含有1个配体,比如1DWD晶体结构(1DWD.pdb),可以直接键入下面命令:
1 | $ pdb2receptor -pdb 1DWD.pdb |
其输出为receptor.oeb.gz,受体结构就准备好了。实际上,1DWD结构里结合配体的名称为MID,你还可以键入如下命令:
1 | $ pdb2receptor -pdb 1DWD.pdb -ligand_residue MID |
其效果与上述的例子是一样的。但是,当PDB结构里含有两个配体的时候,比如1IEP就包含有两个配体,需要指定哪个残基,如果此时按第一种方式键入命令:
1 | $ pdb2receptor -pdb 1IEP.pdb |
会得到下面的提示:
1 2 3 4 | No ligand specified with -ligand_residue. 2 possible ligands detected Auto detected Ligand 1 residues : STI201A : SMILES Cc1ccc(cc1Nc2nccc(n2)c3cccnc3)NC(=O)c4ccc(cc4)C[NH+]5CC[NH+](CC5)C Auto detected Ligand 2 residues : STI202B : SMILES Cc1ccc(cc1Nc2nccc(n2)c3cccnc3)NC(=O)c4ccc(cc4)C[NH+]5CC[NH+](CC5)C To create a receptor pass one of the above residues, or specify -pick_arbitrary_ligand |
此时,我们需要通过-ligand_residue来指定一个配体,比如配体1(STI201):
1 | $ pdb2receptor -pdb 1IEP.pdb -ligand_residue STI201 |
如果不希望采用默认的输出文件名称,可以用-receptor参数来控制输出文件,比如:
1 | $ pdb2receptor -pdb 1IEP.pdb -ligand_residue STI201 -receptor 1iep_receptor.oeb.gz |
这时,得到的输出文件不是默认的receptor.oeb.gz而是1iep_receptor.oeb.gz。
(2) receptor_setup
receptor_setup命令将一个蛋白结构及其共晶的配体或覆盖结合位点的盒子转化为分子对接用的受体文件。与pdb2receptor不同的是,receptor_setup需要将配体-受体复合物拆分成两个文件:一个是受体蛋白结构文件,另一个是共晶的配体文件或box文件。
输入文件的准备:没有特别的地方。如果用共晶的配体来指定结合位点,确保共晶配体从蛋白结构里删除并保存为一个单独文件。
主要的输入文件:
蛋白结构:删除了共晶的配体;
共晶的配体结构:从蛋白复合物结构里提取出来,单独保存为一个文件;
盒子(box):如果不用共晶的配体结构,可以用一个盒子,其覆盖了结合位点;
输出:受体文件
完整的参数如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | Input -protein : File with the structure of a protein. Setup With Bound Ligand -bound_ligand : A file containing, only, the structure of a ligand bound to the active site. Setup With Box -box : File containing a box enclosing the active site. -addbox : Extends the box edges by the specified number of Ångströms. Options -no_strip_water : Do not strip waters from the protein. Output -receptor : Output receptor file. |
以1IEP为例,假设受体结构为protein.mol2,共晶的配体为ligand.mol2,生成对接用受体文件,键入下面命令:
1 | $ receptor -protein protein.mol2 -bound_ligand ligand.mol2 -receptor 1iep_receptor.oeb.gz |
(3) apopdb2receptor
单没有复合物结构,仅有一个无共晶配体的蛋白结构时,我们通过指定一个靠近结合位点的氨基酸来准备受体文件:
1 | $ apopdb2receptor -pdb 3E1X.pdb.gz -site_residue TRP215B |
上面命令以3E1X晶体结构为例, -site_residue TRP215B参数告诉apopdb2receptor以215B号残基附近作为结合位点准备受体结构。
FRED要求配体或化合物数据库预先生成3D多构象文件,典型的方法是用OMEGA对化合物进行构象搜索,保存为配体结构文件。以1IEP的配体STI(Gleevec)为例,其SMILES代码如下:
1 | Cc1ccc(cc1Nc2nccc(n2)c3cccnc3)NC(=O)c4ccc(cc4)C[NH+]5CC[NH+](CC5)C |
将上面一行存为sti.smi的文件,用OMEGA生成多构象文件即可以用于分子对接计算:
1 2 3 | $omega -in sti.smi -out sti.oeb.gz -ewindow 10 \ -maxconfs 1000 -enumNitrogen true \ -enumRing true -prefix sti |
或者再简单点:
1 | $omega -in sti.smi -out sti.oeb.gz |
上述命令得将STI生成3D构象、构象搜索,将低能的构象保存起来,输出到sti.oeb.gz。
注:OMEGA可以处理很多种格式文件,除了上面的SMILES之外,还可以是SDF,MOL,MOL2,PDB等常见格式文件。
现在准备好了受体文件(1iep_receptor.oeb.gz)与配体文件(sti.oeb.gz),FRED分子对接就非常简单了,直接键入下面的命令:
1 | $fred -receptor 1iep_receptor.oeb.gz -dbase sti.oeb.gz |
如果你的机器是多核心的处理器,还支持OpenMPI并行:
1 | $fred -mpi_np 12 -receptor 1iep_receptor.oeb.gz -dbase sti.oeb.gz -prefix sti_fred |
默认情况下,FRED生成几个输出文件。除非你用了其它的输出前缀(-prefix)参数,FRED会以fred为前缀命名所有的输出文件:
| 文件名称 | 描述 |
| fred_docked.oeb.gz | 对接到受体(1iep_receptor.oeb.gz)的数据库(sti.oeb.gz)中打分靠前的500个分子(本例只有一个化合物)。 |
| fred_undocked.oeb.gz | 数据库中(sti.oeb.gz)那些不能对接到活性口袋里的化合物(通常是因为分子太大而放不进结合位点)。如果全部对接成功,则不生成该文件。 |
| fred_score.txt | tab分隔的文本文件,包含有打分最高的500个化合物的名字与分值。 |
| fred_report.txt | 文本文件,报告了对接过程。 |
| fred_settings.param | 对接的参数文件。 |
| fred_status.txt | 记录了对接过程运行状态。 |
FRED完整的参数列表如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 | Execute Options
-param : A parameter file
-mpi_np : Number of MPI processes to launch.
-mpi_hostfile : Path to hostfile to be used for launching MPI processes.
Dock Options
-dock_resolution : Docking resolution
Input
-receptor : Receptor file
-dbase : Multiconformer molecules to be docked
-conftest : Set the test for detecting if sequential molecule records in
the ligand database are conformers.
-molnames : Molecule names file
Output Files
-docked_molecule_file : Output file for docked molecules.
-undocked_molecule_file : Output file for undocked molecules.
-score_file : Outfile names and scores of docked molecules to this text
file.
-report_file : Text file a docking report will be written too.
-settings_file : Write settings for the run to this file.
-status_file : Write periodic status updates to this file.
Output Options
-hitlist_size : Number of top scoring molecules to output. If this flag is
set to 0 all molecules will be outputted as they are docked
(unsorted).
-num_poses : Number of poses to retain for each docked molecule
-annotate_scores : Add VIDA score annotations to docked molecules
-score_tag : Overrides the default tag used to attach the score to the
output molecules
-save_component_scores : Save components of the total score to SD data on
the docked molecules
-no_extra_output_files : Suppress the default output of the score, status,
settings, report and undocked files.
-no_dots : Suppress writing a . to standard error for each docking
molecule (or x in the case of a failure).
-prefix : The text passed to this parameter will be pre-appended all
default output filenames (it does not affect output filenames explicity
set by the users). |
分子对接的结果可以用可视化软件VIDA进行分析;或者第三方软件比如Pymol进行分析;还可以用自带的docking_report生成PDF的报告:
1 2 | $docking_report -docked_poses fred_docked.oeb.gz \ -receptor 1iep_receptor.oeb.gz -report_file fred_docked_report.pdf |
PDF会总结出分子对接(虚拟筛选)命中化合物的关键信息,如图3所示:
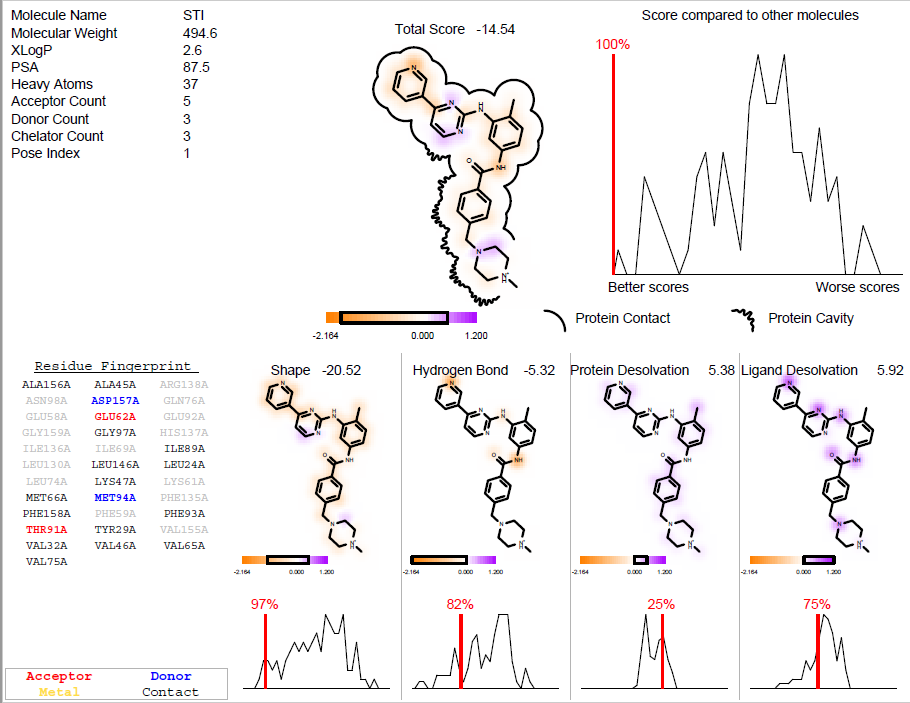
图3. OEDocking的docking_report结果分析报告
四. 总结
抗白血病药物格列卫是ABL激酶抑制剂,1IEP晶体结构为格列卫(STI, GLEEVEC)与ABL激酶的复合物结构。现在,我们用分子对接来预测可能的结合模式,顺带观察计算能否重现结构晶体结构的结合模式。
- 受体结构准备
- 对接计算用配体结构准备
- 对接计算
- 结果分析
从PDB数据库上下载1IEP结构(1iep.pdb.gz),用pdb2receptor命令准备受体文件:
1 | $pdb2receptor -pdb 1iep.pdb.gz -ligand_residue STI201 -receptor 1iep_receptor.oeb.gz |
输入:1iep.pdb (STI与ABL激酶的PDB复合物结构)
输出:1iep_receptor.oeb.gz(用于对接计算用的受体文件)
用Chemdraw、ChemAxon或任意结构式编辑软件画好药物格列卫的结构,保存为SMILES格式文件:sti.smi, 你可以复制下面行,再保存。
1 | Cc1ccc(cc1Nc2nccc(n2)c3cccnc3)NC(=O)c4ccc(cc4)C[NH+]5CC[NH+](CC5)C STI |
现在要用OMEGA对刚化合物的结构进行构象搜索、生成多构象文件,键入如下命令:
1 2 3 | $omega -in sti.smi -out sti.oeb.gz -ewindow 10 \ -maxconfs 1000 -enumNitrogen true \ -enumRing true -prefix sti |
输入:sti.smi (待对接计算的化合物,SMILES格式文件)
输出:sti.oeb.gz (OMEGA生成的多构象文件)
现在用FRED命令开始进行对接计算:
1 | $fred -receptor 1iep_receptor.oeb.gz -dbase sti.oeb.gz |
输入:1iep_receptor.oeb.gz (第一步生成的受体文件)与sti.oeb.gz(第二步生成的配体多构象文件)
输出:六个新文件,其中对接的pose保存在fred_docked.oeb.gz;其中fred_score.txt为打分的结果文件。
默认情况下每个化合物只输出打分最佳的pose,如果需要输出更多,可以设定num_poses选项;打分函数的成份也可以通过save_component_scores选项来打开:
1 | $fred -receptor 1iep_receptor.oeb.gz -dbase sti.oeb.gz -num_poses 20 -save_component_scores true |
用docking_report命令生成相互作用PDF文件:
1 2 | $docking_report -docked_poses fred_docked.oeb.gz \ -receptor 1iep_receptor.oeb.gz -report_file fred_docked_report.pdf |
生成一个新的PDF文件:fred_docked_report.pdf,见图3。
还可以用VIDA或pymol(需要输出为sdf或mol2等格式)以便从3D观察结合模式,或比较计算与实验的差异。下图4展示了打分最高的计算pose与实验结果的差异。
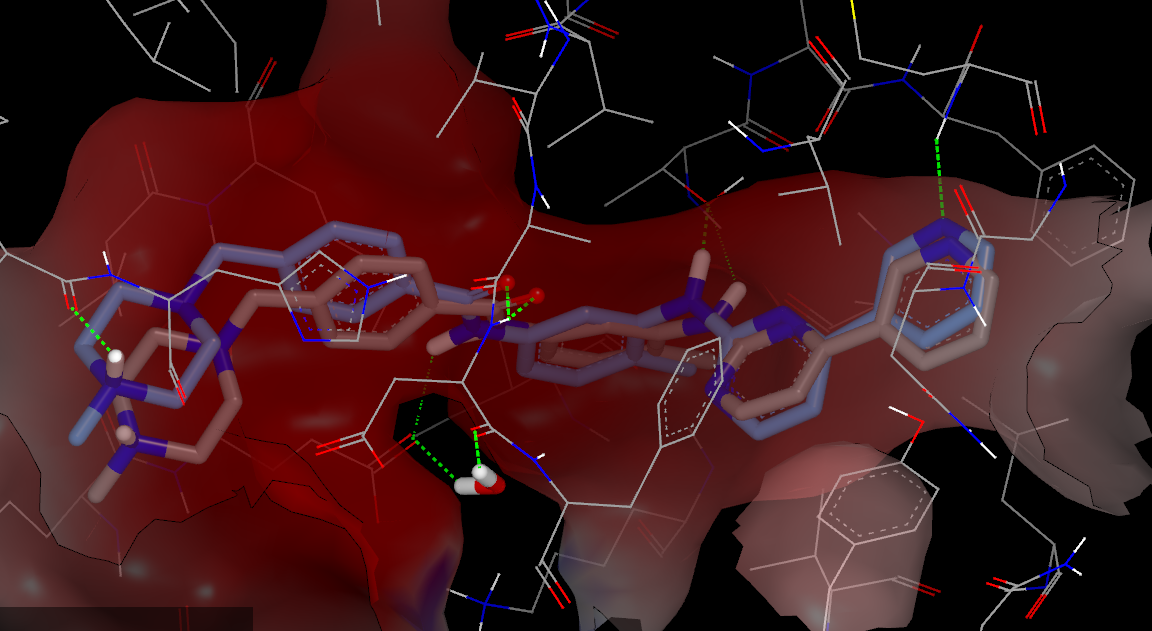
图4. FRED预测的打分最高的pose(灰色)与实验的pose(蓝色),除了哌嗪环位置有点不同,其余的差异很小。
五. 从这里开始可以做什么
- 用FREEFORM进一步考察对接pose稳定性
- 收集各种靶点的受体文件,用于化合物的靶点预测、作用谱预测
见教程:http://blog.molcalx.com.cn/2016/07/02/freeform-conformer-stability.html
五. 文献
(1)McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J Chem Inf Model 2011, 51 (3), 578–596.
(2)Hawkins, P. C. D.; Skillman, A. G.; Warren, G. L.; Ellingson, B. A.; Stahl, M. T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures From the Protein Databank and Cambridge Structural Database. J Chem Inf Model 2010, 50 (4), 572–584.
六. 联系我们获取免费试用
电话:020-38261356
电邮:info@molcalx.com
网站:http://www.molcalx.com.cn
试用要求提供:
姓名:
单位:
地址:
电话:
电邮:
如果是学生,请提供导师的:电话与电邮。


