
摘要:VirtualFlow是一款开源的、基于结构的、超高通量虚拟筛选平台,Gorulla等人成功地用8000个核心花两周时间完成对10多亿化合物进行虚拟筛选,发现了nM水平的KEAP1抑制剂。本文演示了如何在Cloudam上使用VirtualFlow进行此类超高通量虚拟筛选,以VirtualFlow自带的算例VFVS_GK演示了:1)如何准备输入文件;2)编辑配置文件;3)规划计算任务;4)执行虚拟筛选。
作者:李青松,肖高铿
日期:2020/07/05
关于VirtualFlow
基于结构的虚拟筛选是发现先导化合物的重要途经,其发现的苗头化合物的质量随着筛选的化合物数量增加而提高1。虽然已经有很大的化合物库,但是进行这么大规模基于结构虚拟筛选的计算能力一直是个问题,比如进行此类计算集群的可获得性、高效性与灵活性一直是个挑战。
VirtulFlow是哈佛医学院Gorulla等人2,3开发的开源超高通量虚拟筛选平台,它可以让你以巨量并行的方式在计算集群上完成超大规模数据库的虚拟筛选任务。据报道NAF2-KEAP1通路在氧化应激与炎症中起到关键的作用与多种疾病相关,目前已有10个靶向KEAP1的药物处于临床研究阶段,9个药物处于临床前研究阶段。在Gorulla等人4的研究中,用VirutalFlow将13亿的Real化合物库与3.3亿的ZINC化合物库对接到KEAP1-NAF2相互作用界面的KEAP1结合位点,识别出一系列结构多样与KEAP1结合、阻断NAF2-KEAP1相互作用的潜在化合物。生物实验测试结果表明,其中一个化合物与KEAP1的结合亲和力达到nM水平(Kd=144nM)并且阻断NAF2-KEAP1相互作用。
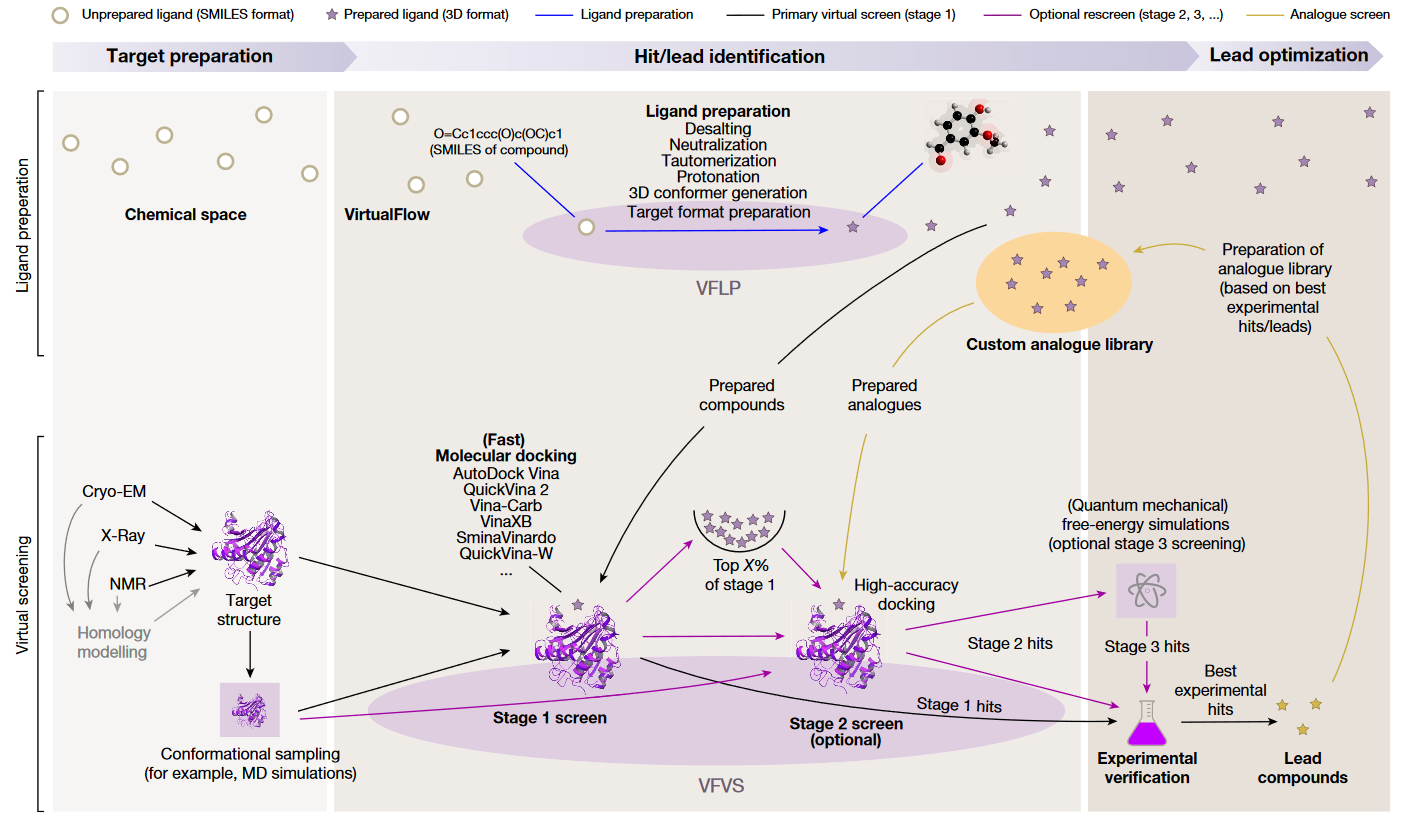
Figure 1. VirtualFlow的虚拟筛选流程
Gorulla的虚拟筛选分两步完成:1)对常规蛋白刚性、配体柔性的策略将13亿Real数据库以及3.3亿ZINC数据库对接到Keap1,取打分最高的300万化合物。本步骤在8000核心的机器上花了4周时间完成。2)用蛋白柔性、配体柔性策略,将上一步打分最高的300万化合物进一步打分,将KEAP1与NAF2相互界面的13残基考虑为构象柔性,用Smina Vinardo与AutoDock Vina进行计算,分别重复两次。对第一步打分最高的98个化合物以及第二步打分最高500个化合物进行了实验验证。
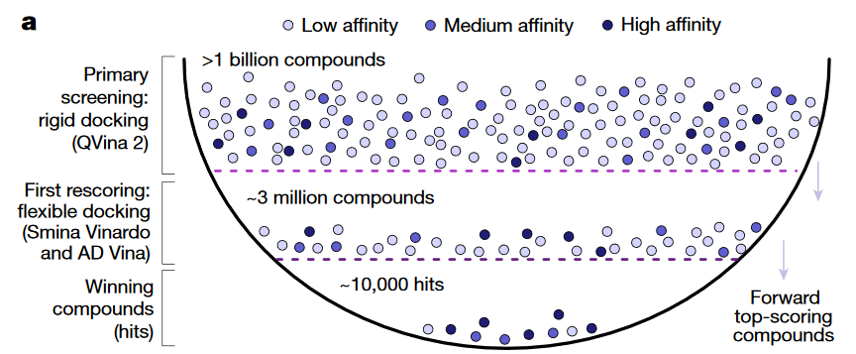
Figure 2. KEAP1的虚拟筛选策略
上述虚拟筛选采用QVINA2分子对接软件进行初筛、再用SMINA与VINA进行两次的蛋白柔性对接打分两次可以通过VirtualFLow的参数文件简单的设置而实现。本文针对这些参数的设置进行演示。
关于云端软件(Cloudam)云E算力平台
Cloudam的云E算力平台整合了全球主流公有云近50个地域的高性能计算资源,开箱即用,无需硬件投入、无需运营维护、无需任务排队,支持一键式提交作业,具备自主学习与深度学习能力,能够根据用户的实例类型与计算要求智能推荐匹配合适的计算机型,将计算的虚拟损耗降至最低,提升计算效率,降低计算成本。
虽然VirtualFlow是开源的,但是将该代码迁移到自己的云服务器上还需要仔细配置与调试、需要一定的学习成本。本着降低学习成本、“开箱即用”的客户服务原则,Clouam的工程师们预先在云端配置好了软件,仅需对少数与项目相关的参数进行设置即可对13亿的Real数据库与3.3亿的ZINC数据库进行基于结构的虚拟筛选。本文将重点演示如何在Cloudam上用VirtualFlow进行基于结构的虚拟筛选。
详细操作步骤
VirtualFlow的文档与教程可以从它的主页获得:https://virtual-flow.org。本文的主要目的是演示如何在Cloudam上使用它。
1. 准备工作
我们假设:
(1)你计划用VirtualFlow自带的13亿Real数据库与3.3亿ZINC数据库进行虚拟筛选,该数据库已经从VirtualFlow下载好可以直接使用(input-files/ligand-library)。你仅需要从VirtualFlow上按空间坐标下载todo.all文件,也就是需要对接化合物所在6维空间坐标,在计算时会根据坐标提取出化合物来进行分子对接计算。
(2)你已经准备好了PDBQT格式的对接用蛋白结构文件:receptors/4no7_prot.pdbqt
(3)你已经准备好了对接计算用的参数文件:scenarios/qvina02_rigid_receptor1/config.txt。
(4)你计划用QVINA2进行1次的蛋白刚性、配体柔性分子对接虚拟筛选。
上述内容已经提供了一个模板,即VirtualFlow的算例VFVS_GK。
2. 登录管理节点
你首先要在cloudam上创建账号,并开启管理节点,然后webssh登录管理节点。
3. 准备输入文件
以VirtualFlow自带的算例VFVS_GS为例,包含输入文件都包含在该文件夹里:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | \---VFVS_GK
| todo.all
| vf.conf
|
+---input-files
| \---ligand-library
| | README.md
| |
| +---GA
| | GACAAD.tar
| | GACAAE.tar
| | GACAAF.tar
| |
| \---HA
| HACABE.tar
| HACABF.tar
| HACACE.tar
|
+---receptors
| 4no7_prot.pdbqt
|
\---scenarios
+---qvina02_rigid_receptor1
| config.txt
|
\---smina_rigid_receptor1
| config.txt |
- input-files
- receptors
- scenarios
- todo.all
- vf.conf
input-files/ligand-library目录存放事先准备好的配体数据库文件。VirtualFlow采用AutoDock Vina系列软件做为分子对接计算程序,此处为保存已经准备好对接用的PDBQT格式的化合物数据库。VirtualFlow提供了两个已经准备好的数据库可以直接使用:1)含14亿化合物的Real数据库;2)含12亿化合物的ZINC15数据库。
对于自己尚未准备过的化合物库,可以用VFLP来准备,或者按照VFVS_GK/input-files/ligand-library的格式自行准备。
注意:并非该文件夹里的所有化合物都用于对接,只有列在todo.all上的那些文件夹里的化合物才被用来对接。
准备好的、可直接用于对接计算的蛋白结构文件,比如4no7_prot.pdbqt。
提供给不同对接软件用的配置文件,比如qvina02_rigid_rceptor01/config.txt,其内容如下:
1 2 3 4 5 6 7 8 9 | receptor = ../input-files/receptors/4no7_prot.pdbqt center_x = -8.654 center_y = 2.229 center_z = 19.715 size_x = 24.0 size_y = 26.25 size_z = 22.5 exhaustiveness = 8 cpu = 1 |
是待对接计算的化合物列表。在input-files/ligand-library里存储的Enamine Real库化合物有14.6亿,但是这些化合物只是准备好可以用来计算,而不必全部都需要被对接计算。只有被列在todo.all里的那些化合物才被计算。以前6行为例:
1 2 3 4 5 6 | GACAAD_00000 9 GACAAF_00000 22 GACABD_00000 27 GACABF_00000 48 GACACC_00000 1 GACACD_00000 30 |
VirtualFlow按化学空间位置管理化合物,空间中相邻的几个化合物被打包压缩在一个文件,第一列对应该压缩文件的名称,第二列对应其中的化合物数。我们需要第二列的数值的总和,来规划后面每节点需处理的化合物数。
vf.conf是Virtualflow的配置文件,是最重要的文件,解释如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 | # Values have to be separated by colons ":" and without spaces, e.g: docking_scenario_names=vina-rigid:smina-rigid # Used for instance for the folder names in which the output files are stored # Should not be changed during runtime, and be the same for all joblines # Settable via range control files: Yes # 对接场景名称,可以任意命名。出于管理方便,建议与对接场景scenario名称一致 # 计算结果输出目录将以此命名,可以进行一个或多个对接计算场景,使用":"隔离 docking_scenario_names=qvina02_rigid_receptor1:smina_rigid_receptor1 # Relative path wrt to the tools folders # In each input folder must be the file config.txt which is used by the docking program to specify its options # If other input files are required by the docking type, usually specified in the config.txt file, they have to be in the same folder # Should not be changed during runtime, and be the same for all joblines # Settable via range control files: Yes # 配置文件config.txt所在目录,每个对接计算需要一个, ../input-files/为固定位置,不可修改,仅需将目录scenarios下的子目录填入即可 docking_scenario_inputfolders=../input-files/qvina02_rigid_receptor1:../input-files/smina_rigid_receptor1 # Possible values: qvina02, qvina_w, vina, smina_rigid, smina_flexible, adfr # Values have to be separated by colons ":" and without spaces, e.g: docking_scenario_programs=vina:smina # The same programs can be used multiple times # Should not be changed during runtime, and be the same for all joblines # Settable via range control files: Yes # 对接计算所用的软件,合法的值为:qvina02, qvina_w, vina, smina_rigid, smina_flexible, adfr docking_scenario_programs=qvina02:smina_rigid # Series of integers separated by colons ":" # The number of values has to equal the number of docking programs specified in the variable "docking_programs" # The values are in the same order as the docking programs specified in the variable "docking_scenario_programs # e.g.: docking_scenario_replicas=1:1 # possible range: 1-99999 per field/docking program # The docking scenario is comprised of all the docking types and their replicas # Should not be changed during runtime, and be the same for all joblines # Settable via range control files: Yes # 每个ligand需要被重复计算的次数 docking_scenario_replicas=1:1 # When the folders are initially prepared the first time, the central todo list will be split into pieces of size <central_todo_list_splitting_size>. One task corresponds to one collection. # Recommended value: < 100000, e.g. 10000 # Possible values: Positive integer # The smaller the value, the faster the ligand collections can be distributed. # For many types of clusters it is recommended if the total number of splitted todo lists stays below 10000. # Settable via range control files: Yes # todo.all文件中按找多少行数进行文件分割处理,建议按照分割后的文件数量在2000一下来设置此参数 central_todo_list_splitting_size=1000 # Used as a limit of ligands for the to-do lists # This value should be divisible by the next setting "ligands_todo_per_refilling_step" # Settable via range control files: Yes # 每个处理进程需要处理的ligand的数量,注:总queque数=申请的总核心数, todo.all中累加为总的待处理ligand数量 # 可以按照每个ligand处理需要的时长 x(乘) ligands_todo_per_queue得出每个轮次需要的时长 # 可以理解为每个核心需要计算的化合物数,全部计算完保存起来,如果计算失败,那么这些化合物没有计算输出 ligands_todo_per_queue=500 # The to-do files of the queues are filled with <ligands_per_refilling_step> ligands per refill step # A number roughly equal to the average of number of ligands per collection is recommended # Settable via range control files: Yes # 单次计算时填充数量 # ligands_todo_per_queue数量的化合物,分几批送进去计算,每次送的化合物数 ligands_per_refilling_step=100 # Not (yet) available for LSF and SGE (is always set to 1) # Should not be changed during runtime, and be the same for all joblines # Settable via range control files: Yes # 应为1 steps_per_job=1 # Sets the slurm cpus-per-task variable (task = step) in SLURM # In LSF this corresponds to the number of slots per node # Should not be changed during runtime, and be the same for all joblines # Not yet available for SGE (always set to 1) # Settable via range control files: Yes # 应为每节点的核心数 cpus_per_step=4 #steps_per_job * cpus_per_step = 使用的总核心数 #steps_per_job=625, cpus_per_step=16,表示启动16核的机器共625台,总共10000核 |
上述输入文件的组织可以下载模板,然后进行修改或替换即可:
1 2 | cd ~ cp /public/software/.local/easybuild/software/virtualflow/examples/VFVS_GK/vf.comf . |
现在用我们事先准备好的受体文件、docking参数文件与todo.all文件,根据前面提到的VFVS_GK文件结构,替换对应内容就可以了。
4. 虚拟筛选
为了方便计算,我们准备了creatworkflow.sh脚本,将该脚本复制到HOME目录,
1 2 | cd ~ cp /public/software/.local/easybuild/software/virtualflow/vfvs/createworkflow.sh . |
并按需要修改前面两行:
1 2 | DOCKERING_CONFIG_DIR=/home/cloudam/VFVS_GK DOCKERING_WORKING_DIR=/home/cloudam/myfirsttest |
其中第1行是我们刚刚准备的输入文件目录;第2行是进行虚拟筛选的工作目录,你可以任意命名,这里我命名为myfirsttest。
还需要修改最后一行:
1 | ./vf_start_jobline.sh 1 1 templates/template1.slurm.sh submit 1 |
将第2个参数设定为节点数。比如,我需要用500个节点计算,则修改为:
1 | ./vf_start_jobline.sh 1 500 templates/template1.slurm.sh submit 1 |
现在可以运行该脚本开始虚拟筛选:
1 2 | cd ~ ./creatworkflow.sh |
5. 结果查看
在tools目录提供了结果统计工具可以快速浏览结果。比如:
1 2 | cd ~/myfirsttest/tools ./vf_report.sh -c vs -d qvina02_rigid_receptor1 |
结果如下图3所示,可以看到化合物的打分分布情况。
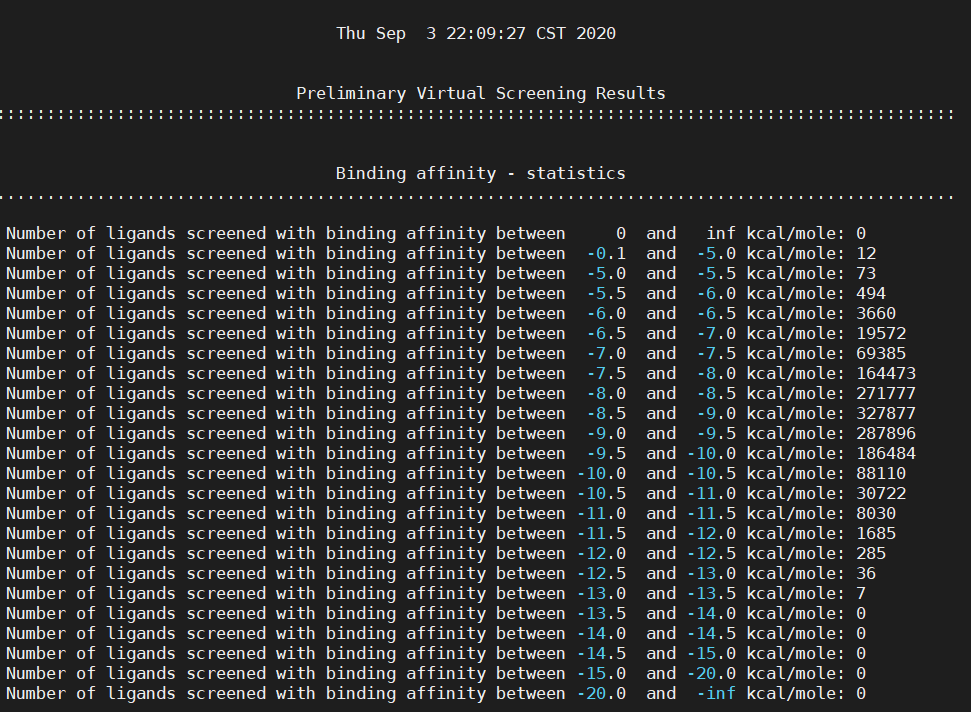
Figure 3. Virtual Flow的报告生成
如图3所示,最后一列给出了对接打分取值范围内的化合物数,比如打分为-12.0~-12.5kcal/mol的化合物为285个。
6. 结果排序
需要将VFTools复制一份到本地:
1 2 3 | cd ~/myfirsttest cp -r /public/software/.local/easybuild/software/virtualflow/vftools/VFTools . export PATH=../../VFTools/bin:$PATH |
在VirtualFlow计算完毕,还可以对所有的化合物进行排序,进入myfirsttest目录,创建专用的目录,键入命令:
1 2 3 | cd ~/myfirsttest mkdir -p pp/ranking cd pp/ranking |
对所有的化合物排序:
1 | vfvs_pp_ranking_all.sh ../../output-files/complete/ 2 meta_tranche |
如果不能正常使用,请键入:
1 | vfvs_pp_ranking_all.sh -h |
根据帮助,确保参数都是正确的。同时确认已将VFTools/bin加入到PATH环境变量。
给出打分最佳前100个化合物的列表,保存为compound:
1 | head -n 100 qvina02_rigid_receptor1/firstposes.all.minindex.sorted.clean > compounds |
7.提取打分最靠前100个化合物的对接结合模式(pose)
创建新的目录,用来保存pose
1 2 3 | cd .. mkdir -p docking_poses/qvina02_rigid_receptor1 cd docking_poses/qvina02_rigid_receptor1 |
根据第6步的列表(componds),将top 100化合物的pose提取出来:
1 2 | cp ../../ranking/compounds . #复制列表到当前目录 vfvs_pp_prepare_dockingposes.sh ../../../output-files/complete/qvina02_rigid_receptor1/results/ meta_tranch compounds dockingsposes overwrite |
生成三个文件,两个目录:
1 | compounds compounds.energies compounds.energies.uniq.csv dockingsposes dockingsposes.plain |
每个打分最佳的pose已经按排名顺序以PDB格式保存在dockingsposes.plain目录里,可以直接用可视化软件对结果进行分析,推荐用FlareViewer做为可视化软件对结果进行分析。
关于VirtualFlow的数据库与计算列表(todo.all)
VirtualFlow提供了两个数据库可以下载:Enamine的Real Library与ZINC15 library,分别有14.6亿与12亿化合物。像Real或ZINC15库等这么这种超大型的化合物库,进行虚拟筛选都花费巨大:预计需要15-20万元,这相当昂贵,因此有偏向地选择部分化学结构进行虚拟筛选是有必要的。
Virtual flow提供了六维化学空间坐标定位帮你从巨大的化学空间中选择部分你感兴趣的化合物:
- Molecular weight (MW)
- Partition coefficient (SlogP)
- Number of hydrogen bond acceptors (HBA)
- Number of hydrogen bond donors (HBD)
- Number of rotatable bonds (RotB)
- Topological polar surface area (TPSA)
通过鼠标调整紫色圆圈的位置,可以设定每个坐标的取值范围,完成对感兴趣选择,如下图4所示:
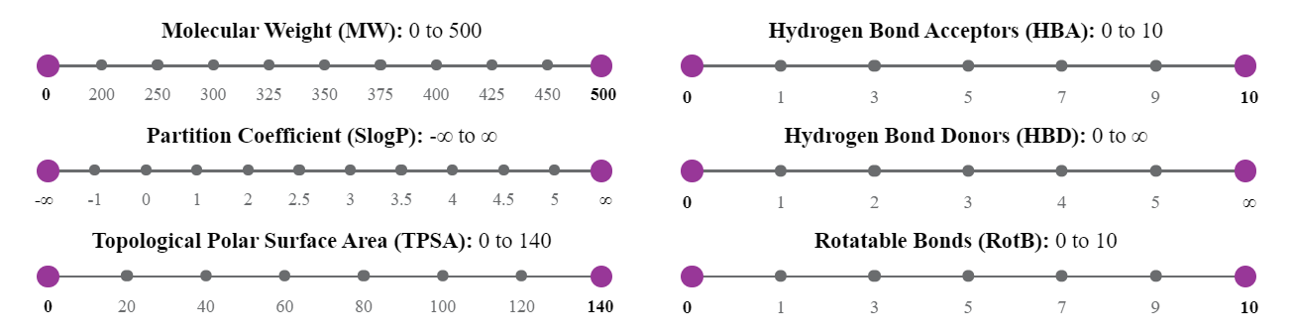
Figure 4. 6维化学空间定位
然后,如图5所示,在Download method for tranches的下拉菜单里选择一种你熟悉的下载方法,我推荐wget,然后点击旁边的Download按钮。这时,你将下载到的是Real library下载脚本;将该文件放在input-file/ligand-library目录,在该目录执行脚本就可以下载到直接可用的数据库。点击Collection-length file旁边的Download按钮,则下载到todo.all文件,该文件就是需要对接的化合物列表。

Figure 5. 下载
蛋白结构准备
蛋白结构准备需要考虑狠多方面的问题,在《教程 | 蛋白结构的检查与准备》一文中已经有详细讨论。这里推荐用Flare或OpenEye/Spruce用来进行蛋白结构准备,然后保存为PDB或mol2以备后面进一步格式转化为vina系列软件可用的格式。
也可以用Pymol与openbabel来进行结构准备,参见教程:Cloudam教程 | AutoDock Vina分子对接软件的使用。
化合物结构准备流程
以下流程供大家参考:
- 标准化
- 脱盐
- 中和
- 过滤
- 去重复结构
- 质子化状态枚举
- 互变异构体枚举
- 立体化学枚举
- 3D结构生成
- 格式转化
不同来源的化合物结构,其SMILES编码千差万别,需要统一表示。以钠盐为例,一个常见的表达方式为钠与羧酸氧之间通过键连接,比如:
1 | [Na]OC(=O)c1ccc(C[S+2]([O-])([O-]))cc1 |
这不利于后续的脱盐处理(会被当成金属有机化合物而不是盐),需要标准化为非键连接:
1 | [Na+].O=C([O-])c1ccc(CS(=O)=O)cc1 |
如果你没有其它可靠的工作做化合物标准化与验证的话,我推荐你使用用MolVS[5,6],具体方法参见Standardizing a molecule章节
我们只需要保留酸根与碱基部分,其它的部分不需要。比如上一步的分子,我们将Na+去除,只保留酸根部分:
1 | O=C([O-])c1ccc(CS(=O)=O)cc1 |
如果你没有可靠的其它软件帮你做脱盐处理,我推荐RDKit或MayaChemTools[7]的RDKitRemoveSalts.py。
通常可选,因为接下来马上要进行“滴定”,取特定pH值下的质子化状态。但是中和之后的结构有利于下一步的去重复结构操作。
过滤掉不想要的化合物很重要,合理的过滤可以避免不必要的计算。RDKit与前面提到的MayaChemTools可以帮忙进行各种各样的过滤处理。
同一个活性成分的不同盐通常被当成不同的结构,脱盐之后就可能出现重复结构,因此有必要进行去重处理。
RDKit与前面提到的MayaChemTools的RDKitRemoveDuplicateMolecules.py可以用于去重复结构处理。
根据研究靶标的结合位点特征,枚举特定pH值下的化合物质子化状态是很重要的。比如,对于含有金属的结合位点,该金属与化合物发生配位键相互作用。那么准备这样的配体的时候,不妨取其在高pH值时的质子化状态,以保证该化合物有机会与结合位点的金属发生配位相互作用。比如磺胺在高pH时N上带负电,可以与蛋白Zn发生配位键。但是质子化中性的磺胺,就不能与Zn发生配位作用了。
如果你没有直接可用的软件处理质子化状态,openbabel的oababel选项p可以处理特定pH下质子化状态。
不同的互变异构体与蛋白发生相互作用的形式不同,分子对接结果也不同,因此包含对的互变异构体是很重要的。
前面提到的MolVS可以帮你枚举互变异构体,参见API:MolVS-tautomer。
当SMILES或SDF文件含有双键、手性中心等立体化学因素时,有时存在立体化学没有限定清楚的问题,这是需要对其进行枚举以保证包含正确的结构。
RDKit可以用来枚举化合物的立体化学,参见教程:RDKit进行立体化学枚举。
最关键的一步是3D结构的生成,已经有很多高质量的软件可以用来生成化合物的3D结构。恨过软件比如OMEGA,corina在这一步可以实现前面的部分工作。
Vina系列软件需要用PDBQT格式的问价作为输入文件,如果你生成的3D结构文件不是PDBQT格式,可以用MGLTools或openabel进行格式转化。
OpenEye等商业软件包含了上面化合物结构准备所需全部工具。OpenEye的QUACPAC与OMEGA提供了上面所需的化合物结构准备全部功能。
视频演示
根据上面的流程,进行了些许的改编,演示了虚拟筛选的操作过程。
相关主题
- OpenEye | 化合物数据库的准备
- VirtualFlow教程 | Enamine数据库的更新及其虚拟筛选
- Flare Viewer: 免费、高质量的药物设计视图工具
- 将sdf转化为virtualflow的tranches样式
该文详细描述了如何用OpenEye准备小分子数据库,得到的sdf文件,我们提供了python脚本转化为virtualflow样式。
介绍了在Cloudam上可获得的两个现货数据库:Enamine的Adv与HTS。并将createworkflow.sh拆解为单个命令行,以详解Virtual flow的虚拟筛选流程。
用于结果分析,不需要任何命令行。
文献
- Lyu, J.; Wang, S.; Balius, T. E.; Singh, I.; Levit, A.; Moroz, Y. S.; O’Meara, M. J.; Che, T.; Algaa, E.; Tolmachova, K.; Tolmachev, A. A.; Shoichet, B. K.; Roth, B. L.; Irwin, J. J. Ultra-Large Library Docking for Discovering New Chemotypes. Nature 2019, 566 (7743), 224–229. https://doi.org/10.1038/s41586-019-0917-9.
- Gorgulla, C. VirtualFLow https://github.com/VirtualFlow (accessed Jul 19, 2020).
- Gorgulla, C. VirtualFlow. https://www.virtual-flow.org (accessed Jul 19, 2020).
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.-F.; Fischer, P. D.; Coote, P. W.; Padmanabha Das, K. M.; Malets, Y. S.; Radchenko, D. S.; Moroz, Y. S.; Scott, D. A.; Fackeldey, K.; Hoffmann, M.; Iavniuk, I.; Wagner, G.; Arthanari, H. An Open-Source Drug Discovery Platform Enables Ultra-Large Virtual Screens. Nature 2020, 580 (7805), 663–668. https://doi.org/10.1038/s41586-020-2117-z.
- MolVS: Molecule Validation and Standardization. https://molvs.readthedocs.io
- MolVS: Molecule Validation and Standardization. https://github.com/mcs07/MolVS
- MayaChemTolls. http://www.mayachemtools.org
- Open Babel: The Open Source Chemistry Toolbox. http://openbabel.org
联系我们
使用VirtualFlow,请前往Cloudam注册账号。


