
摘要:从PDB数据库下载的蛋白结构并不完美,正确准备与否影响了分子对接的结果。本文介绍了如何在分子对接前检查蛋白的结构,并加以适当的准备。
一. 前言
从PDB数据库下载的晶体结构只是一个能与实验数据拟合的模型,而并非实验数据本身。而基于的结构的分子对接对蛋白的结构高度敏感,因此仔细审查下载的蛋白结构、并正确地准备就显得非常重要。本文以MLL1为例子来说明如何检查下载的蛋白结构、并进行结构准备。实际上,不只是分子对接,基于结构的药效团识别也需要正确准备蛋白结构才能识别出正确的相互作用及药效团。
MLL1是跟血癌相关的致病蛋白,它通过将辅酶SAM(S-adenosyl methionine)的甲基转移到组蛋白(Histone)上而影响基因的转录, 而辅酶本身也被转化为SAH(S-adenosyl-L-homocysteine)。SAM是非常柔性的分子,它通过不同的构象与不同的酶结合。SAM的结合位点被视为具有成药性,已经有人着手研究治疗癌症与神经方面疾病的药物[1]。
二. 蛋白结构的检查与准备
我们的任务
我们要完成的任务是将SAM的产物SAH对接回结合位点,检查能否重现结合模式。
SAH的SMILES:O=C(O)[C@@H](N)CCSC[C@H]3O[C@@H](n2cnc1c(ncnc12)N)[C@H](O)[C@@H]3O
图1. SAH的结构
我们的挑战
SAH的结合模式预测并不容易,因为面临着几个挑战:
- 如图1所示,SAH含有7个柔性键。
- 具有柔性的糖环:如果一开始环是错误的构象,对接后能否得到正确的结合模式呢?
- 结合位点呈半开放状态
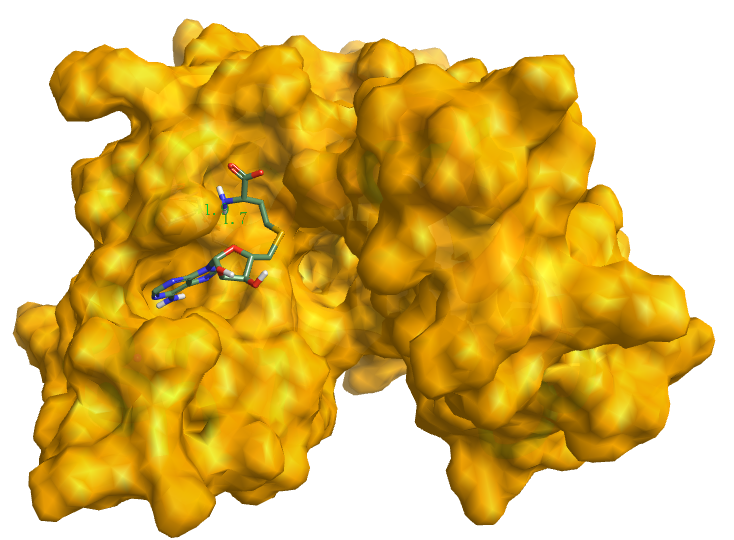
图2. SAH的结合位点,PDB code: 2W5Y
我们需要重点做的事
- 检查蛋白结构
- 配体质子化状态处理
- 配体的原子类型、键的类型检查
- 配体环的柔性处理
因为侧链酰胺的羰基氧(=O)与-NH2具有同样的质量,单晶X-衍射实验不能分辨O与-NH2的位置,因此侧链酰胺的结构可能是错误的。检查PDB 2W5Y结合位点里的ASN3906,如图3所示,发现ASN3906的酰胺取向对配体结合不利(图3位置2),也与临近残基GLU3939的相互作用不利(图3位置3)。因此该基团的结构是错的,因此需要纠正这个酰胺基团。纠正后ASN3906如图6所示,不仅可以与配体产生氢键相互作用,还可以与旁边的GLU3939发生氢键作用。
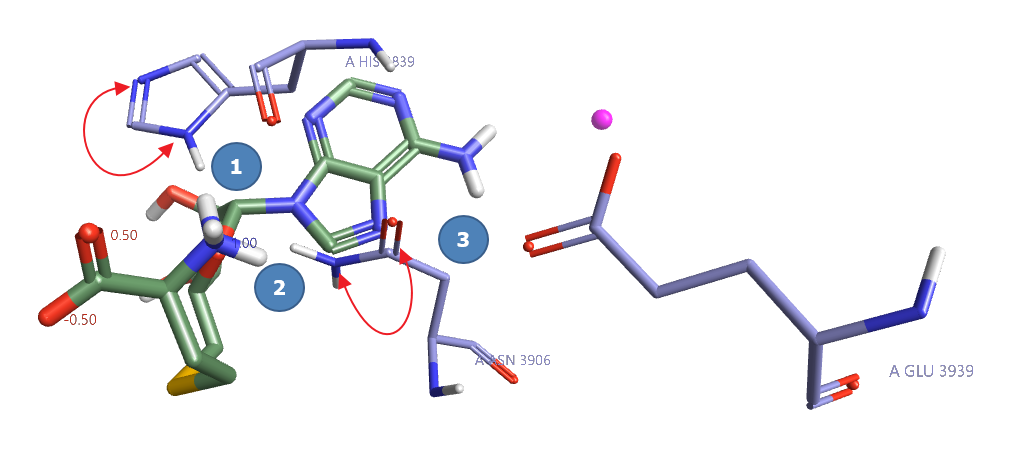
图3. 初始的蛋白结合位点结构(PDB:2W5Y):随机加氢而不考虑氢键方向则(1)位置的氢有问题;此外,侧链酰胺的取向经常也是错误的(2,3位置需要互换)。
HIS的咪唑具有三种质子化状态与互变异构体,如图4所示,咪唑环质子化的氮可以在Delta位,也可以在Epsilon位,或者两个位置都处于质子化状态。单晶X-衍射实验不能确认氢原子的位置,因此不能分辨HIS应该是那种质子化状态或互变异构体形式,因此需要仔细的设定。如图3所示,HIS3839咪唑是Delta位质子化,指向配体的质子化N正电中心,这并不合理。HIS3839咪唑合理的质子化位置应该使得咪唑与配体-NH3发生氢键作用才是能量有利的。
|
|
|
|
图4. HIS咪唑的质子化状态与互变变异构体
图5. SAH的质子化状态
SAH具有一个羧基与胺基,羧基处于去质子化状态而胺基处于质子化状态。如果不知道正确的质子化状态,应当对质子化状态(以及互变异构体)进行枚举。
在这个例子中不存在原子类型或键的类型错误,但是建议仔细检查原子类型与键的类型,常用的软件基本都有方便的编辑工具进行编辑。比如在FLARE中可以鼠标右键选需要编辑的配体、然后edit就可以。
注意:PDB数据库提供的配体2D结构是自动注释的,并非原作者提供,也非人工提供,经常在结构上是错的,建议阅读原文以确认正确的结构。比如PDB 3I65注释的配体结构质子化状态与原文就不一致。
糖的环是柔性的,而大部分分子对接软件在对接计算中环按刚性处理,此时需要在对接前将环的构象进行枚举;如果对接软件支持环的构象搜索,则可以开启环柔性处理功能而忽略环的初始构象。
三. 详细的操作流程
3.1 蛋白结构的自动准备(Flare)
很多结构准备软件都具备自动蛋白结构准备与配体结构准备的功能,比如LeadFinder可以按指定的pH值加氢、对氢的取向进行结构优化;考虑复合物结构里配体的质子化状态与互变异构体状态。2W5Y结构经LeadFinder在pH7条件下准备之后,结合位点的结构满足我们的要求,如图6所示。
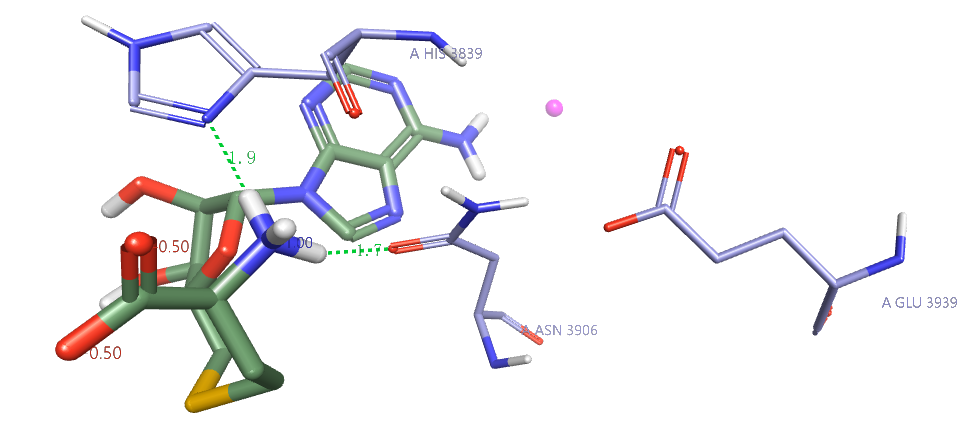
图6. 经LeadFinder准备过的MLL1-SAH的复合物结构
具体的操作流程请参见《Flare 教程 | 分子对接-结合模式预测》
如果发现配体结构有质子化状态、原子类型或键的类型错误,需要用鼠标右键点击对应的配体,选择Edit,可以对结构进行编辑,修改至正确后保存、退出编辑状态。
3.2 蛋白结构的手动检查与准备
无论您用的软件有多么的聪明,我都强烈建议您去检查一下蛋白结构。这里以Ligandscout为例来说明如何检查蛋白结构,其它的软件也是同样的道理。Ligandscout提供了“Analyze functional groups”可以列出需要检查的蛋白侧链,并且提供了原子与键的编辑器可以手动修改原子类型、键的类型、修饰配体等等,当然也可以用来修改质子化状态与互变异构体。
- 首先下载2W5Y结构,点击黄色盒子,点击Create Pharmacophore生成药效团。
- 官能团分析
- 选择需要编辑的原子或键,用原子与键编辑器对互变异构体或质子化状态进行编辑
- 编辑完毕,点击Create Pharmacophore观察相互作用
- 分子对接计算
Molecules > Analyze functional groups,此时在3D视窗会出现Amide,Imidazole的Label。这些label对应的氨基酸残基就是我们需要去重点检查的对象。
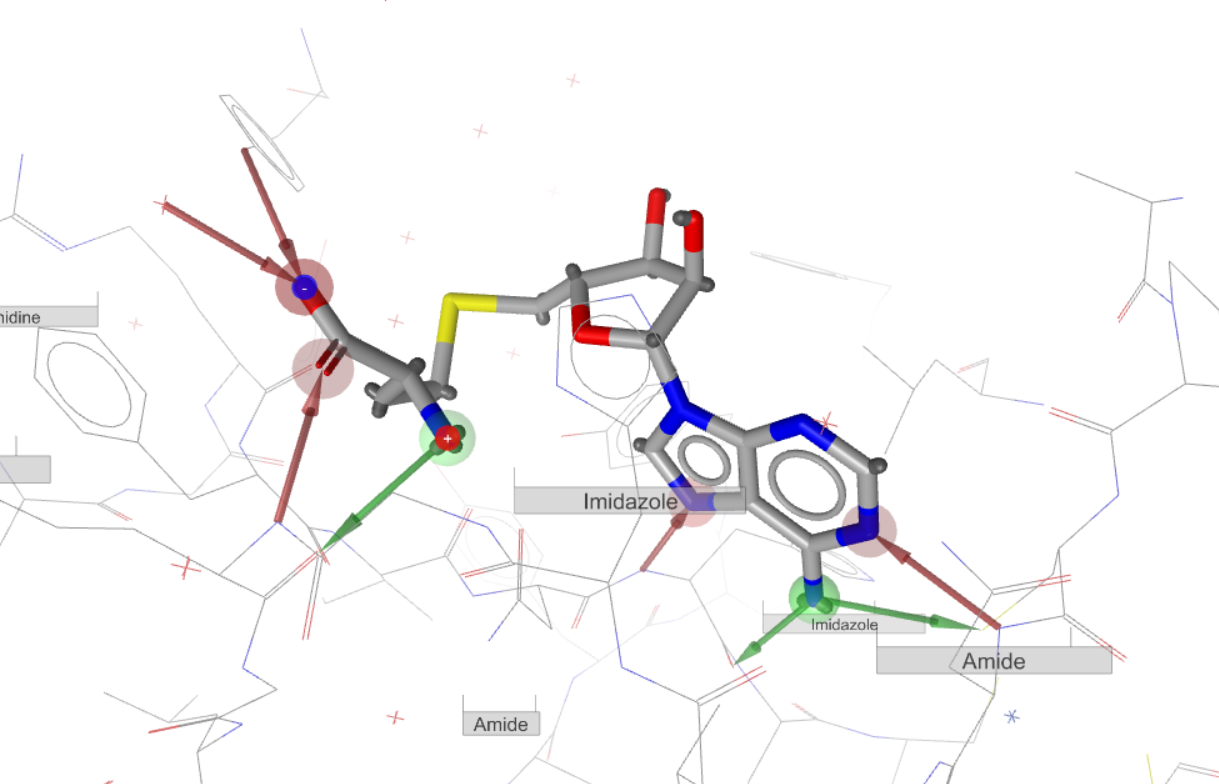
图7. 官能团分析给出了配体周围存在质子化状态、互变异构可能的氨基酸残基
从图7可以就看到,有两个氨基酸残基靠近配体-NH3正电中心,这两个残基本来应该与-NH3形成氢键相互作用,但是药效团分析并没有看到该相互作用,说明结构有误。
比如,我们需要将ASN3906的侧链酰胺N与氧换个位置,第一步先将原子进行替换,再将键进行替换:以O替换为N为例,先用鼠标单击O使之处于被选中状态(图8步骤1),再点击”Change Atom type按钮”(图8步骤2),从元素周期表上选择N(图8步骤3),则将O替换为N了。用同样的方式,将N替换为氧。
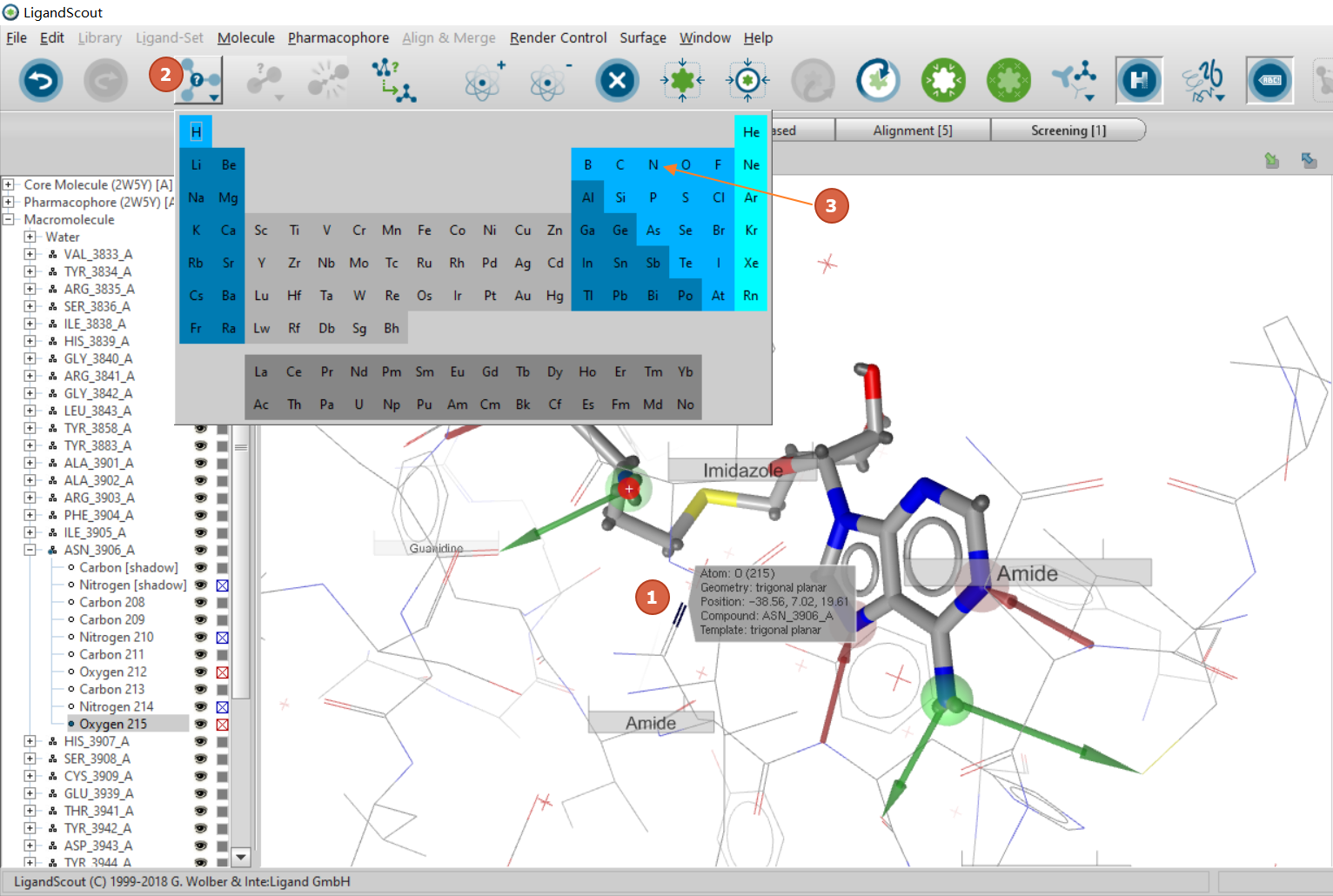
图8. 原子类型的替换
为了方便观察,我们将ASN3906展示为球-棍状:先双击ASN3906的任意一个原子以选中整个残基(图9步骤1),再点击“Render Control”菜单里的“Select”(图9步骤2),选择“Ball-and-Stick”(图9步骤3),完成渲染。
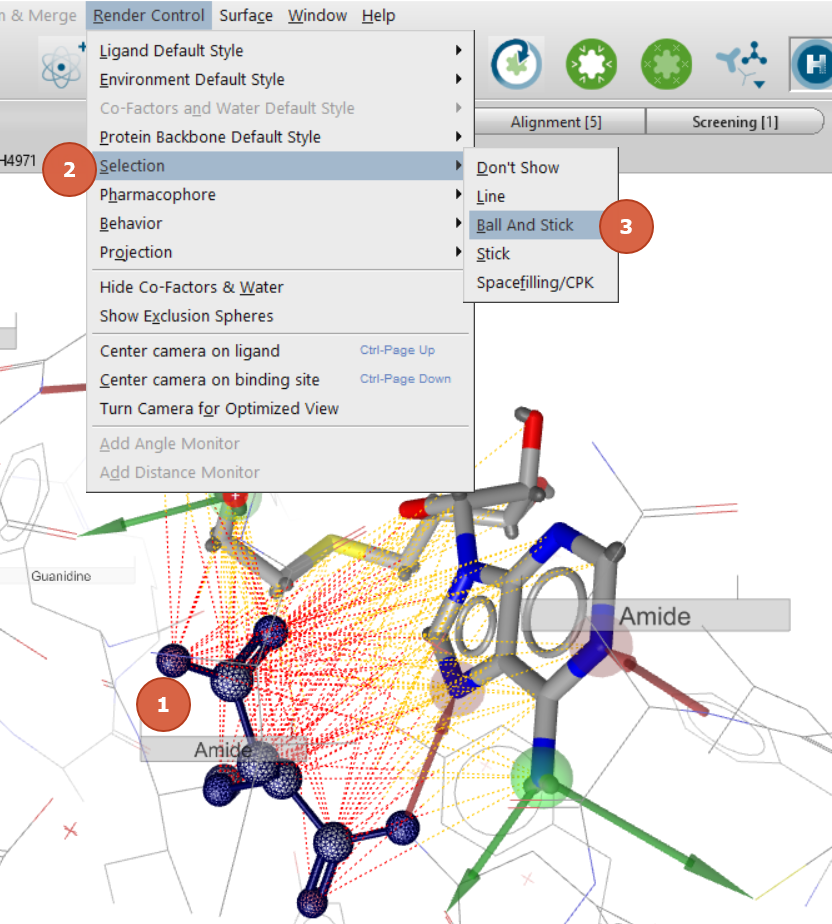
图9. 对被选中的原子或残基进行渲染
原子类型编辑好之后,可以修改键的类型:鼠标单击要修改的键(图10步骤1),点击”Change Bond type”(图10步骤2),,再从菜单选择需要的键的类型(图10步骤3)。
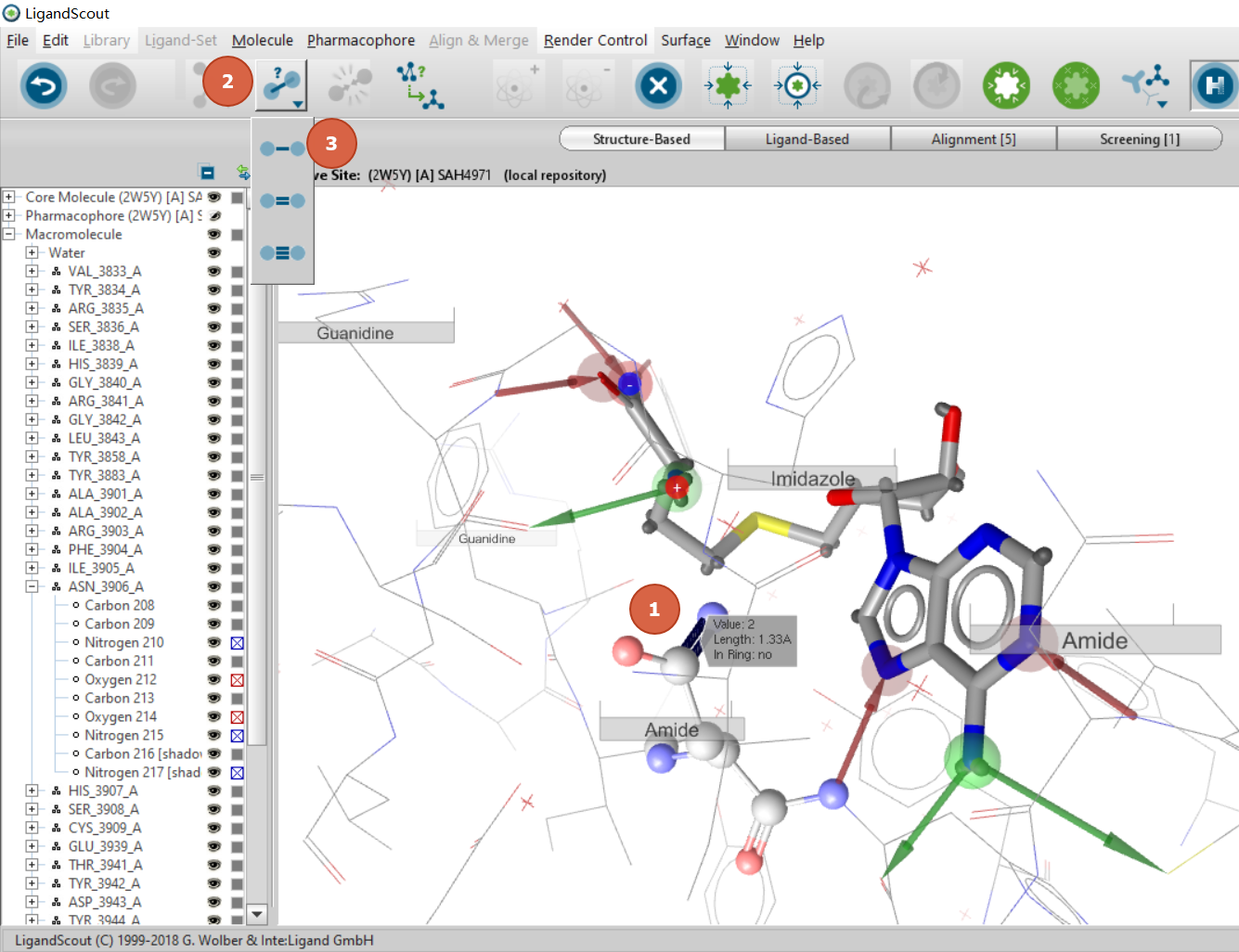
图10. 编辑键的类型
如果相互作用不满意,继续编辑直到满意为止。最后,我们会发现,新的药效团模型多出了与ASN3906以及HIS3839的两个氢键相互作用,见图11与12。
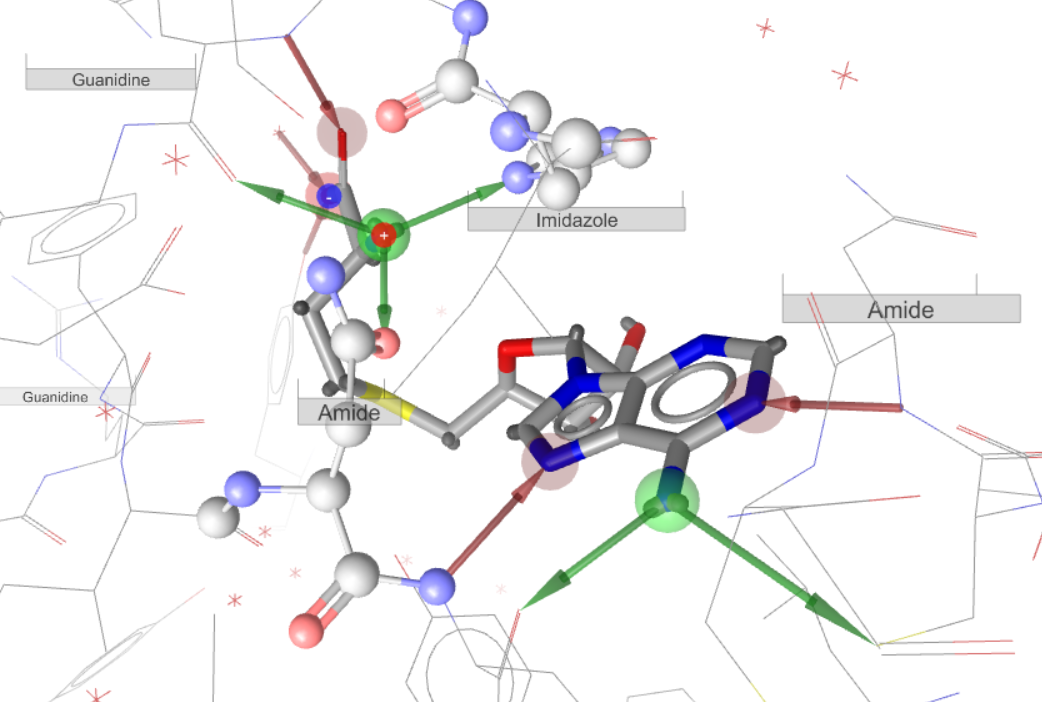
图11. 2W5Y修正蛋白结构后的相互作用模式3D图(Ligandscout生成)
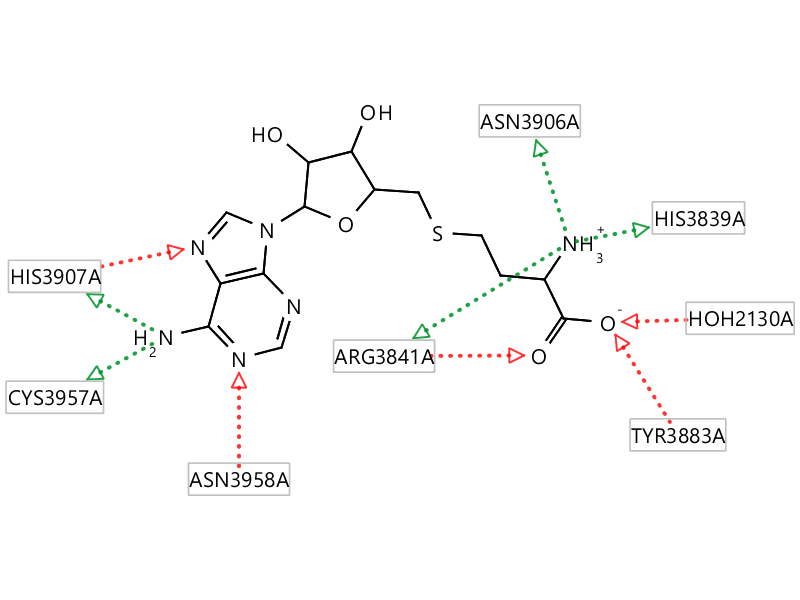
图12. 2W5Y修正蛋白结构后的相互作用模式2D图(Ligandscout生成)
现在蛋白结构已经准备完毕,可以将待对接计算的分子导入Ligandscout进行分子对接计算,具体流程见:《Ligandscout教程-VINA分子对接》
3.3 配体结构的准备
按照图5的质子化状态、立体化合物准备结构,存为SMILES、Mol2或SDF格式。这里我们提供了一个难度比较大的初始结构,其中环不是复合物2W5Y的糖构象以作为配体环预处理的练习来使用,还提供了由Ligandscout/icon生成的25个低能构象。
SAH的3D结构下载,其中糖的环不同于复合物结构(随机构象):SAH.mol2
用Ligandscout/icon生成25个低能构象(icon构象):SAH_icon.sdf
3.4 分子对接结果
- 对接方法:Ligandscout/Vina + 蛋白结构准备 + 随机构象
- 对接方法:Ligandscout/Vina + 蛋白结构准备 + icon构象
- 对接方法:LeadFinder + LeadFinder蛋白结构准备 + 随机构象
- 对接方法:LeadFinder + LeadFinder蛋白进行准备 + icon构象
- 对接方法:Ligandscout/Vina + 不进行蛋白结构准备 + icon构象
Ligandscout/Vina以Vina打分第二的pose重现出了结合模式,该pose被Ligandscout/binding affinity打分排名第一。总的来说,Ligandscout/Vina重现了结合模式,Binding Affinty将正确结合模式识别出来,并排名第一。与复合物里的配体构象相比,除了环的构象外,其余部分基本完全重合,见图13。无论结合位点里的水是否去除,vina都能重现结合模式。
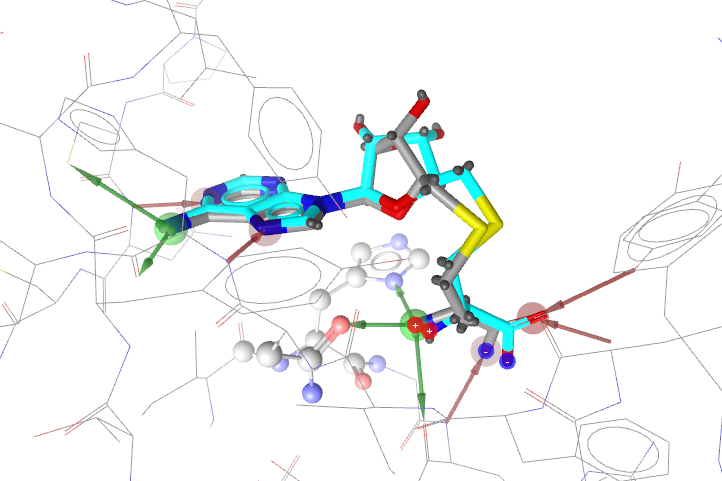
图13. Ligandscout/Vina预测的结合模式:灰色为实验结构,蓝色为预测结构,形状相似性0.84
取一个icon生成的构象进行结合模式预测,结果发现计算结果完全与实验结果重复,环部分也完全匹配(形状相似性0.94),见图14。Vina score与Binding Affinityscore排名第一的构象就是最佳的构象。无论结合位点里的水是否去除,vina都能重现结合模式。
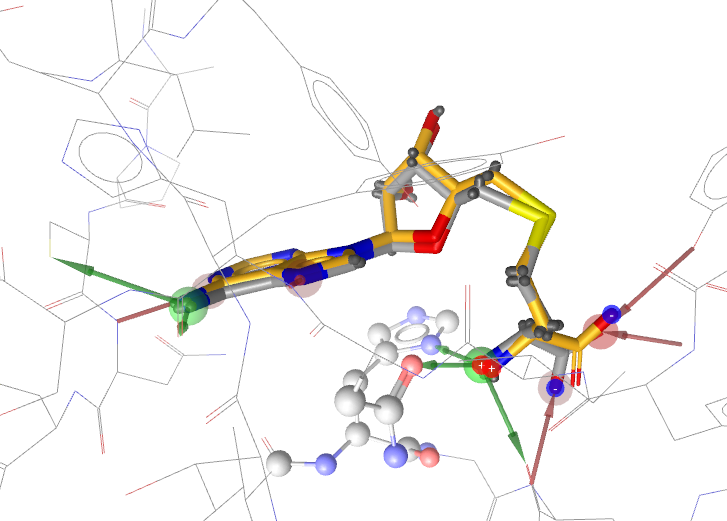
图14. Ligandscout/Vina预测的结合模式:灰色为实验结构,黄色为预测结构,形状相似性0.94
当去除结合位点里的水分子后,LeadFinder不能重现结合模式;而保留水分子后,docking的最佳pose与结合模式pose形状相似性为0.65,但是配体的氨基没有重现结合模式的相互作用(见图15)。
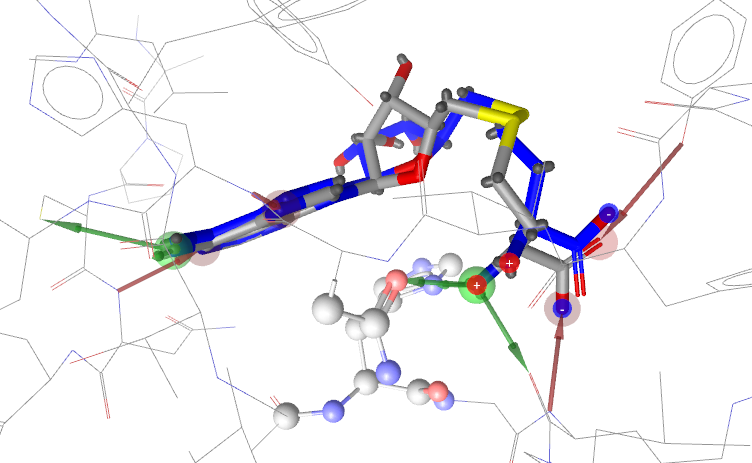
图15. LeadFinder预测的结合模式:灰色为实验结构,蓝色为预测结构,形状相似性0.65
当去除结合位点里的水分子后,LeadFinder不能重现结合模式;而保留水分子后,LeadFinder可以重现结合模式(见图16)。
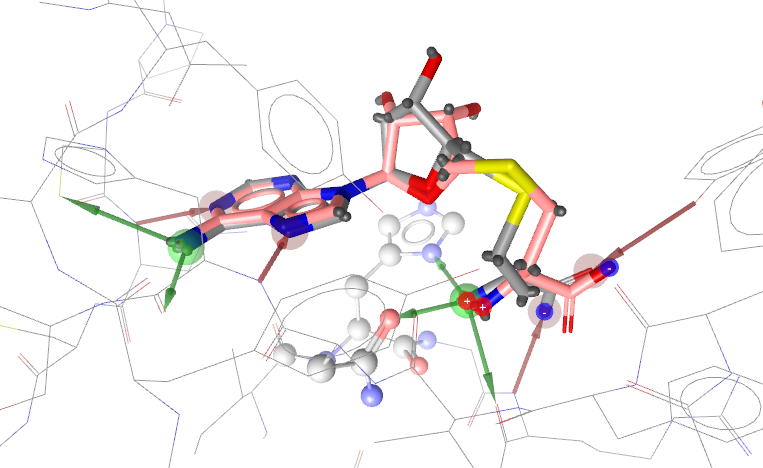
图16. LeadFinder预测的结合模式:灰色为实验结构,粉红为预测结构,形状相似性0.8
去除结合位点里的水分子后,不修复蛋白结构,用icon生成的构象进行结合模式预测,结果发现配体的氨基正电中心部分取向完全错误(图17),不能重现实验结果。
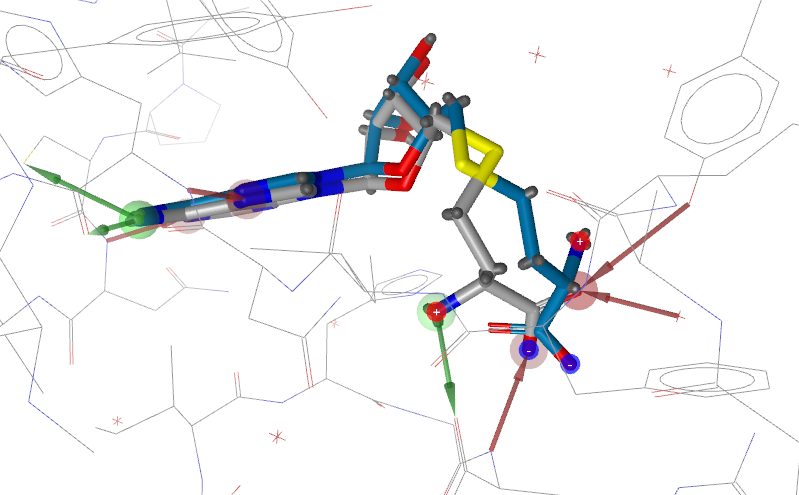
图17. Ligandscout/Vina预测的结合模式:灰色为实验结构,蓝灰色为预测结构,注意:配体-NH3的取向
3.4 小结
- PDB下载的晶体结构也会有”错”
- HIS需要重点检查
- 本文还没有谈到其它方面的内容
- 比较了修复HIS质子化状态与ASN结构错误对分子对接计算结果的影响
常见的有侧链酰胺的取向错误,所以要检查结合口袋里的ASN与GLN侧链结构。在本2W5Y的例子中,我们发现ASN3906是错误的,并演示了在Ligandscout里通过原子与键的类型编辑来纠正此类错误。
X-Ray实验不能确认氢原子的位置,而HIS有3个质子化状态或互变异构体,默认的质子化形式可能就是错误的,通过与配体或周围残基的氢键相互作用可以选择最有利的质子化形式。在2W5Y结构里,结合位点里的HIS3839默认的质子化状态是错误的,通过Ligandscout的原子与键类型编辑可以获得自己需要那个质子化状态或互变异构体形式。
蛋白结构的准备不只是涉及到侧链酰胺与HIS的检查与修正,还有水分子的处理、缺失原子或侧链的修复等等。其中水分子可以通过OpenEye的SZMAP或FLARE的3D-RISM来分析研究,而缺失原子一般的分子软件包都可以修复,残基缺失或一段残基缺失还可以通过同源建模来修复。
我们发现修复与不修复结果差异非常大,检查与修复是非常必要的。
四. 其它需要注意的蛋白结构准备事项
- PDB下载的蛋白结构是否有电子密度数据?
- 结合位点是否已知?
- 配体的结构对了吗?原子类型与键的类型对了吗?键长与键角是否正确?
- 配体是否存在多种质子化状态与互变异构体?
- 观察到预期的相互作用模式了吗?如果没有,是否可以合理解释?
- 蛋白侧链是否可以移动?每个侧链(尤其长侧链的残基)的位置是否都有电子密度数据支持?
- 口袋里是否有水分子或缓冲剂分子?
- 残基缺失或原子缺失
建议确认结合位点里的配体、关键的侧链是否获得电子密度的支持。
确认你要对接的口袋位置。
前面提到过要核对PDB给出的结构与文献2给出的结构不一致的3i65,如下图18所示。
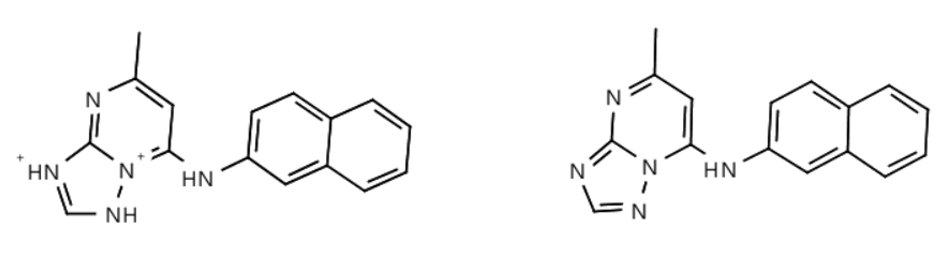
图18. 左边是PDB 3i65给出的2D结构(错误),右边是原始文献结构(正确)
并非所有的“复合物”结构都是复合物结构,有的结构并不体现配体-蛋白的相互作用模式。
对分子对接来说,残基缺失或原子缺失并非必须修复。之所以缺失是因为该位置的原子更容易“动”,得不到电子密度的支持,温度因子高。这也意味着该缺失原子或残基是非常柔性的,可以被其它原子在空间中替换掉(比如被配体“挤”到一边),因此不修复也可以: 以便让配体有机会占据该空间位置。
五. 文献
- Arrowsmith, Cheryl H., et al. “Epigenetic protein families: a new frontier for drug discovery.” Nature Reviews Drug Discovery (2012). 11, 384-400.
- Deng X, Gujjar R, El Mazouni F, et al. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem. 2009. doi:10.1074/jbc.M109.028589.


