
摘要:本文以(+)-鸡蛋花素的绝对构型确证为例介绍了如何通过Flare 构象搜索与Flare QM进行ECD光谱计算,具体步骤包括:XED力场构象搜索、半经验与DFT构象优化、构象去重、激发态计算以及ECD图谱绘制。结果表明,Flare构象搜索与QM计算获得了正确的低能构象系综,并将(+)-鸡蛋花素的绝对构型正确地归属为(1R,5S,8S,9S,10S)-鸡蛋花素,并得到文献结果的验证。此外,Flare比Crest、RDKit、OpenBabel表现出更好的构象搜索性能。Flare不仅是可靠的、综合性的药物设计平台,而且也是可靠的结构确证工具。
肖高铿/2024-04-23
前言
鸡蛋花素(Plumericin)是一种具有抗真菌与抗菌活性的环烯醚萜类天然产物,该分子具有5个手性中心,于20世纪60年代被首次分离。如图1所示,(+)-鸡蛋花素具有两个对映异构体,2007年Stephens等人1用DFT理论计算了它们的VCD、ECD与OR图谱,并与实验图谱进行比较,确证了绝对构型。
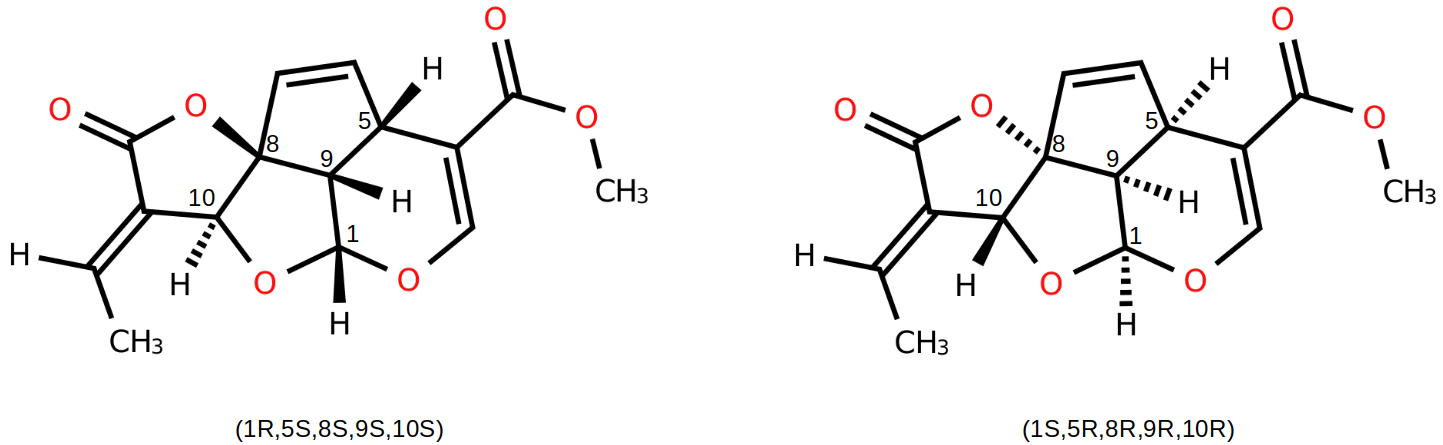
图1. (+)-鸡蛋花素的两个对映异构体
本文的目的是,以Stephens等人1报道的(+)-Plumericin结构确证为例,来演示如何结合Flare2的构象搜索、Flare QM、以及其它量子化合物计算工具来实现ECD图谱计算、确认化合物的绝对构型。整个过程包括下面步骤:
- 用Flare/Conf Hunt在XED力场水平进行构象搜索
- 用Flare QM在xTB-GFN2理论水平进行构象优化
- 构象去重
- 用Flare QM在DFT理论水平进行构象优化
- ECD图谱计算
方法与结果
构象搜索
(1R,5S,8S,9S,10S)-鸡蛋花素的初始结构从PubChem下载(见附件),导入到Flare之后,如图2所示,用Flare |3D-Pose | Conf hunt & Align对(1R,5S,8S,9S,10S)-鸡蛋花素进行构象搜索。在构象搜索与叠合对话框,选择Very Accurate and Slow模式对化合物进行构象搜索。
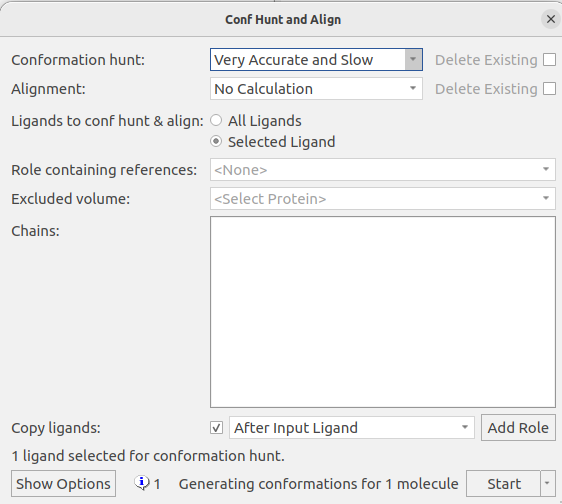
图2. (1R,5S,8S,9S,10S)-鸡蛋花素的构象搜索
点击Show options,设定如下参数:
- Maximum number of conformations: 1000
- No. of High-T Dynamics runs for flexible rings: 20
- Gradient cutoff for conformer minimization: 0.100 kcal/mol/A
- Filter duplicate conformers at RMSD: 0.50 Å
- Energy window: 30.0 kcal/mol
- Acyclic secondary amide handling: Allow amides to spin
- Remove boats and twist-boats: uncheck
- Turn off Coulombic and attractive vdW forces: uncheck
其中酰胺键允许旋转,但是在本次计算里该设置是无效的,因为不涉及这样的酰胺键。将Turn off Coulombic and attractive vdW forces这一选项去掉勾选,也就是启用长程的静电与vdW吸引相互作用。此外,为了得到尽可能多的初始构象,还将能量窗从3kcal/mol调高到30kcal/mol。
在这个计算里面,我们得到一个能量窗为13.5kcal/mol的构象系综,含有11个构象。导出构象系综,保存为SDF格式:xed-ensemble.sdf。
在xTB-GFN2理论水平进行初步的构象优化
如图3所示,使用Flare |3D-Pose | QM | minimize conformations,在xTB-GFN2理论水平对化合物的构象系综进行优化。

图3. 在xTB-GFN2理论水平进行初步的构象优化
计算完毕,导出构象系综,保存SDF格式:xTB-ensemble.sdf。
将SDF格式的构象系综转化为xTB风格的构象系综XYZ文件
用OpenBabel3对结构的进行格式转化:
1 2 3 4 5 6 7 8 | # 用QM_ENERGY对构象排序 sdsort -n -fQM_ENERGY xTB-ensemble.sdf >> ensemble_sorted.sdf # 将SDF名称修改能量值(QM_ENERGY) RDKitSetSDFTitle.py ensemble_sorted.sdf ensemble_energy.sdf QM_ENERGY # 转化为xyz格式,注释行为能量值 obabel -isdf ensemble_energy.sdf -oxyz -O ensemble.xyz |
上述这么处理是为了获得一个xTB风格的构象系综xyz文件,用于后续的构象去重。
构象去重
构象去重可以方便地使用Flare/pyflare的python脚本实现2,也可以借助crest的子命令cregen来实现4,5,6:
1 | crest CONF_1.sdf --cregen ensemble.xyz -chrg 0 -rthr 0.125 -ewin 30 |
其中,CONF_1.sdf是起始的一个xTB-GFN2水平优化过的构象,可以从ensemble.xyz里拿出一个;chrg是体系的net formal charge;ewin设置能量窗,默认为6kcal/mol;rthr设置RMSD,默认为0.125 Ang。
计算完毕,得到聚类后的文件:crest_ensemble.xyz。
在本研究中,11个构象都得到保留,XED与xTB-GFN2水平计算的能量如图4所示,其中energy列是XED力场计算的相对构象能,QM Relative Energy列是xTB-GFN2计算的相对构象能。

图4. 在XED与xTB-GFN2理论水平计算相对构象能
在DFT理论水平单点能计算,去除高能构象
在Flare QM里对上一步骤xTB-GFN2理论水平优化的构象在wB97X-D/def2-TZVP理论水平进行单点能计算,结果如图5所示,其中energy列是XED力场计算的相对构象能,QM Relative Energy列是在wB97X-D/def2-TZVP//xTB-GFN2xTB-GFN2理论水平计算的相对构象能。
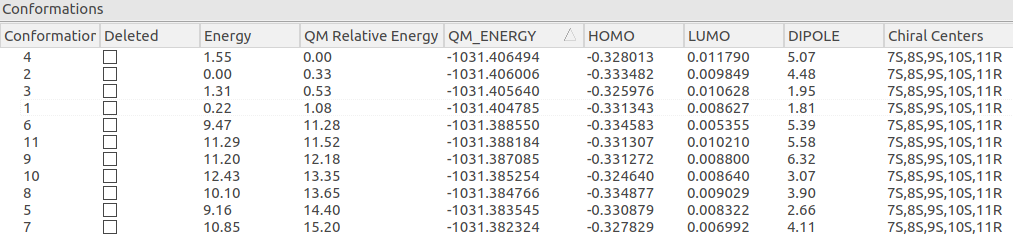
图5. 在wB97X-D/def2-TZVP//xTB-GFN2理论水平计算相对构象能
假设在wB97X-D/def2-TZVP//xTB-GFN2xTB-GFN2理论水平计算的构象能与DFT优化后计算的构象能会发生60%的偏差,那么我们取前面最低能的4个构象也足以覆盖99%以上的构象空间。
在DFT理论水平进行几何优化
在Flare QM里用BP86-D3BJ/DEF2-TZVP理论水平对上一步保留下来的4个构象进行几何优化,并在同样理论水平下使用氯仿(chloroform)作为溶剂进行单电能计算,得到新的构象系综,结果如图6所示。其中energy列是XED力场计算的相对构象能,QM Relative Energy列是在BP86-D3BJ/DEF2-TZVP/IEFPC-Chloroform理论水平几何优化后的相对构象能,QM Energy列是电子结构能量(包含了溶剂效应)。
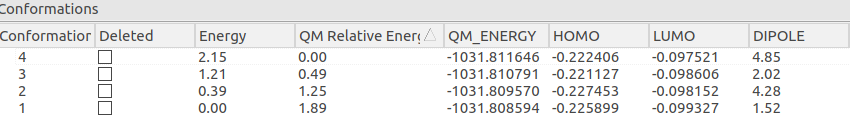
图6. 在BP86-D3BJ/DEF2-TZVP理论水平进行几何优化的电子结构能量与相对能量
优化后的(1R,5S,8S,9S,10S)-鸡蛋花素有四个构象:甲酸甲酯相对于相邻的C=C键有σ-顺式或反式两个取向;含氧原子的五元环有两种取向。这四个构象的关键两面角及其电子结构能量、波尔兹曼权重如表1所示。
表1. 鸡蛋花素理想两面角与构象能
| 构象 | C5-C4-C12-O14 | C8-C9-C1-O11 | ΔE(kcal/mol) | 波尔兹曼权重 |
|---|---|---|---|---|
| 4/a | 180° | -20° | 0 | 0.638 |
| 3/b | 0° | -20° | 0.537 | 0.262 |
| 2/c | 180° | +20° | 1.303 | 0.073 |
| 1/d | 0° | +20° | 1.915 | 0.026 |
注:ΔE是根据电子结构能量计算;两面角的值只是用来表示取向,并非实际值。
图7展示了在BP86-D3BJ/DEF2-TZVP理论水平进行几何优化后的四个构象及其关键的两面角原子编号。其中原子编号不是根据化学信息学而来,而是根据与文献一致为原则下的编号。四个构象的关键两面角与波尔兹曼权重与Stephens等人1报道的基本一致,重现了文献结果。
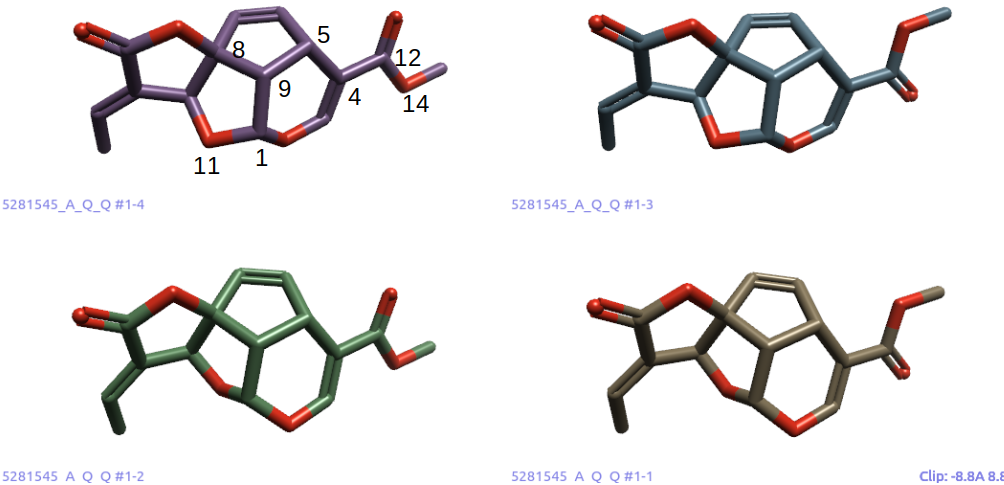
图7. 在BP86-D3BJ/DEF2-TZVP理论水平进行几何优化后的四个构象
总的来说,经过Flare的XED力场构象搜索、Flare QM的xTB-GFN2半经验QM几何优化与结构去重、DFT水平的几何优化,重现了文献报道的四个关键的低能构象。接下来可以在此构象基础上进行光谱模拟。
此外,使用Crest4-6、OpenBabel实现的Confab17、以及RDKit18实现ETKDGv2等几个流行的构象搜索对(1R,5S,8S,9S,10S)-鸡蛋花素生成构象系综,结果表明这些方法不能同时生成a、b、c、d四个关键构象。在我的一个测试中(见补充部分),Crest而只能搜索到b、d构象;OpenBabel与RDKit仅搜索到a、b构象,如表2所示。这说明Flare的构象搜索有其独到的地方,并得到真正的低能构象系综,覆盖了关键构象,而这是图谱计算最重要的步骤。
表2. 不同方法重现文献四个关键构象的结果统计
| 构象 | ΔE(kcal/mol) | Flare | Crest | OpenBabel | RDKit |
|---|---|---|---|---|---|
| a | 0 | \(\checkmark \) | \(\checkmark \) | \(\checkmark \) | |
| b | 0.537 | \(\checkmark \) | \(\checkmark \) | \(\checkmark \) | \(\checkmark \) |
| c | 1.303 | \(\checkmark \) | |||
| d | 1.915 | \(\checkmark \) | \(\checkmark \) |
\(\checkmark \):表示包含该构象
ECD图谱计算
(+)-鸡蛋花素实验ECD光谱在氯仿溶液中进行,包含了表3所示的特征峰1。
表3. (+)-鸡蛋花素实验ECD光谱的特征峰
| 波长(nM) | 旋光强度(10-40esu2cm2) |
|---|---|
| 192 | +15 |
| 215 | -5 |
| 237 | +13 |
| 268 | -0.13 |
溶剂:氯仿
正确模拟实验ECD光谱需要对四个构象的ECD计算图谱进行波尔兹曼加权平均。分别对每个构象都运行至少20个态的TD-DFT计算,ECD光谱预测作为激发态计算的一部分自动完成,其中每一个计算的激发态产生一个旋光强度。用TD-DFT激发态计算ECD图谱可以使用多个软件来实现,比如之前演示过的Flare QM内置PSI49,10与Gaussian11,12,你可以根据这两个教程来计算ECD图谱,除此之外还可以用NWChem13、TURBOMOLE14、ORCA15与AMS16等等。
在这里,我以丰度最高的构象a(CONF_04.xyz)为例,使用ORCA在B3LYP/DEF2-TZVP理论水平进行激发态计算,输入文件如下:
1 2 3 4 5 6 7 | %PAL NPROCS 24 END !B3LYP DEF2-TZVP CPCM(Chloroform) %TDDFT NROOTS 30 TDA FALSE END * xyzfile 0 1 CONF_04.xyz |
四个构象的ECD图谱通过波尔兹曼加权平均得到最终图谱,如图8所示,随着波长的增加,理论光谱和实验光谱的系列峰,尤其是特征峰可以一一对应上:分别在192、215、237、268nM的波长处有对应的有正、负、正、负Cotton效应。真正的图谱比较应该将计算图谱与实验图谱进行拟合,不仅在波长上可能需要进行偏移处理,而且在峰高上进行缩放。由于没有实验图谱的数据点文件,这里只能进行定性地大致比较。
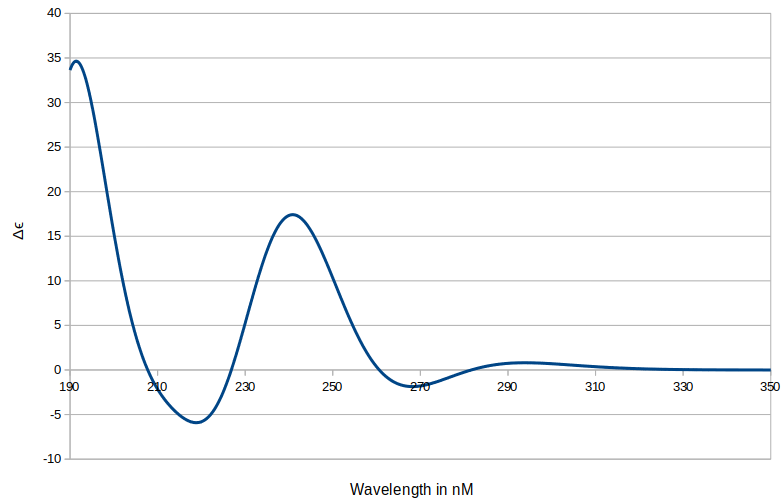
图8. 四个构象波尔兹曼加权平均的ECD图谱(向短波长方向偏移7nm)
我们的计算(1R,5S,8S,9S,10S)-鸡蛋花素ECD图谱与实验的(+)-鸡蛋花素ECD图谱拟合较好,说明(1R,5S,8S,9S,10S)-鸡蛋花素是(+)-鸡蛋花素的绝对构型。
旋光度(Optical Rotation,OR)的计算
(+)-鸡蛋花素在以钠光谱D线(波长为589.3nm)为入射光时测得的比旋光度在+170~+201之间1。PSI410、Gaussian 1611、Turbomole14、AMS16等常见的QM工具可以用来计算特定入射光的比旋光度。比如,此前我们演示过如何用Gaussian 16计算分子的比旋光度19。在B3LYP/DEF2-TZVP理论水平下,用IEFPCM溶剂化模型隐式考虑氯仿的效应,计算的结果如表4所示。
表4. (1R,5S,8S,9S,10S)-鸡蛋花素比旋光度计算值与实验值比较
| 构象 | [α]D | 权重 |
|---|---|---|
| a | 244.88 | 0.638 |
| b | 225.33 | 0.262 |
| c | -50.18 | 0.073 |
| d | -92.96 | 0.026 |
| 波尔兹曼加权平均 | 209.2 | |
| 实验值 | +170~+201 |
溶剂:氯仿
计算的[α]D=209.2,与报道的(+)鸡蛋花素的[α]D实验值+170~+2011非常一致,比旋光度的计算结果支持ECD数据建议的绝对构型指认。同时也可以发现,如果仅使用RDKit与OpenBabel搜索到的构象a、b或者Crest搜索到的构象b、d来计算比旋光度,则与实验值相去甚远,这也进一步说明了合理构象系综的重要性,以及Flare构象搜索的可靠性。
VCD图谱计算
ORCA支持VCD计算,借助Cai•factor,可以非常方便的进行图谱比较,具体操作过程参见前文《用ORCA计算VCD图谱进行绝对构型的指认》,这里不再叙述。
结论
本文以(+)-鸡蛋花素的绝对构型确证为例介绍了如何通过Flare 构象搜索与Flare QM进行ECD光谱计算,具体步骤包括:XED力场构象搜索、半经验与DFT构象优化、构象去重、激发态计算以及ECD图谱绘制。结果表明,Flare构象搜索与QM计算获得了正确的低能构象系综,并将(+)-鸡蛋花素的绝对构型正确地归属为(1R,5S,8S,9S,10S)-鸡蛋花素,这与计算比旋光度的结论是一致的,并得到文献的验证。此外,Flare比Crest、RDKit、OpenBabel表现出更好的构象搜索性能。Flare不仅是可靠的、综合性的药物设计平台,而且也是可靠的结构确证工具。
补充:用Crest、OpenBabel、RDKit进行构象搜索
用Crest进行构象搜索
在真空中用Crest4,5,6(Version 3.0)对从PubChem下载的鸡蛋花素进行构象搜索:
1 | crest plumericin.sdf --gfn2 -chrg 0 -T 24 |
结果发现,Crest生成了4个构象,但是缺少了两面角C5-C4-C12-O14=180°的构象,如图9所示。
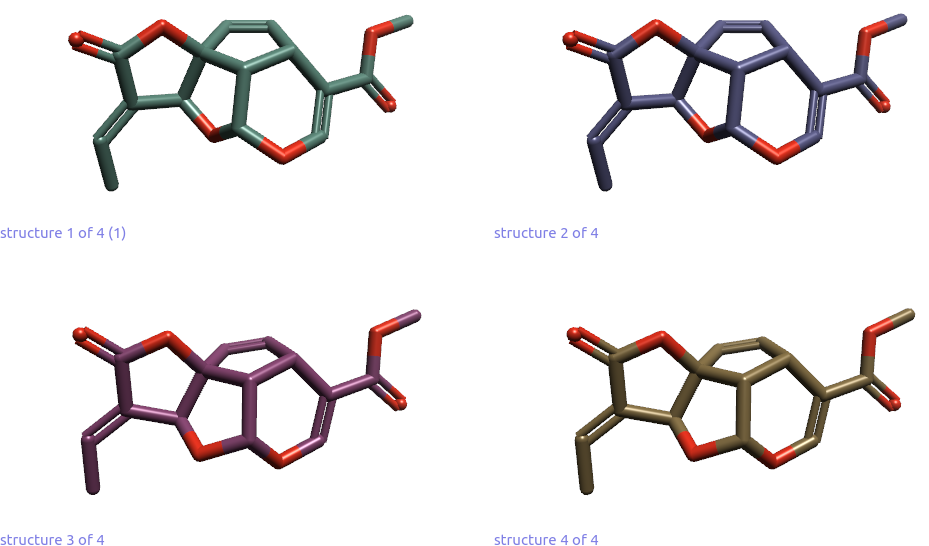
图9.Crest搜索到的4个(+)-鸡蛋花素构象
检查这4个构象,可以发现构象1、2仅是在甲酸甲酯的末端甲基构象存在差异;构象3、4也是同样的情况。因此,Crest本质上仅搜索到Stephens等人报道1b、d构象,而没有发现构象a与c。更重要的是,其中a是全局最低能构象、c的构象能也低于d。因此,在这个算例里,crest生成构象系综未能包含真正的最低能构象。
OpenBabel构象搜索
Confab (Version 1.1.0)是OpenBabel内置的一个系统构象搜索算法17,默认生成最多1百万构象,用来定义重复构象的RMSD截断值为0.5,能量窗设为50kcal/mol:
1 | obabel -isdf plumericin.sdf -osdf -O ob_confab_ensemble.sdf --confab --rcutoff 0.5 --ecutoff 50 |
检查结果,发现总共生成了两个构象,分别是Stephens等人1报道的a、b构象。
RDKit构象搜索
RDKit是流行的化学信息学软件18,在本研究中,使用AllChem包的ETKDGv2构象生成器,以RMSD=0.5为重复构象截断值,MMFF94s力场对构象进行优化并评估构象能,设置能量窗50kcal/mol,输出最多200个构象。结果发现RDKit共生成两个构象,分别为Stephens等人1报道a、b构象。
附件
从PubChem下载的初始plumericin结构:plumericin.sdf
Flare构象搜索结果:xed-ensemble.sdf
参考文献
- Stephens PJ, Pan JJ, Devlin FJ, Krohn K, Kurtán T. Determination of the absolute configurations of natural products via density functional theory calculations of vibrational circular dichroism, electronic circular dichroism, and optical rotation: the iridoids plumericin and isoplumericin. J Org Chem. 2007 Apr 27;72(9):3521-36. doi: 10.1021/jo070155q. Epub 2007 Mar 28. PMID: 17388636.
- Flare™, Cresset®, Litlington, Cambridgeshire, UK. https://www.cresset-group.com/flare
- OpenBabel. https://github.com/openbabel/openbabel
- CREST. https://github.com/grimme-lab/crest
- CREST Docs. https://crest-lab.github.io/crest-docs
- CREST — A program for the exploration of low-energy molecular chemical space, Pracht, P.; Grimme, S.; Bannwarth, C.; Bohle, F.; Ehlert, S.; Feldmann, G.; Gorges, J.; Müller, M.; Neudecker, T.; Plett, C.; Spicher, S.; Steinbach, P.; Wesołowski, P.A.; Zeller, F.; J. Chem. Phys., 2024, 160, 114110. DOI: 10.1063/5.0197592
- Introduction to CENOS. https://xtb-docs.readthedocs.io/en/latest/CENSO_docs/censo.html
- Stefan Grimme, Fabian Bohle, Andreas Hansen, Philipp Pracht, Sebastian Spicher, and Marcel Stahn. Efficient Quantum Chemical Calculation of Structure Ensembles and Free Energies for Nonrigid Molecules. The Journal of Physical Chemistry A 2021 125 (19), 4039-4054. DOI: 10.1021/acs.jpca.1c00971
- 用Flare QM计算化合物的ECD图谱. http://blog.molcalx.com.cn/2022/09/27/flare-ecd.html
- PSI4: Open-Source Quantum Chemisty. https://psicode.org
- Gaussian 16. https://www.gaussian.com
- Gaussian教程–ECD计算. http://blog.molcalx.com.cn/2016/05/23/gaussian-ecd-tutorial.html
- NWChem: Open Source High-Performance Computational Chemistry. https://www.nwchem-sw.org
- TURBOMOLE: Program Package For Electronic Structure Calculations. https://www.turbomole.org
- ORCA 5.04. https://orcaforum.kofo.mpg.de
- AMS. https://www.scm.com/amsterdam-modeling-suite
- O’Boyle, N.M., Vandermeersch, T., Flynn, C.J. et al. Confab – Systematic generation of diverse low-energy conformers. J Cheminform 3, 8 (2011). https://doi.org/10.1186/1758-2946-3-8
- RDKit Version 2023.3.02. https://www.rdkit.org
- Gaussian教程 | 计算分子的比旋光度. http://blog.molcalx.com.cn/2019/12/17/gaussian-optical-rotation.html.


