
用计算方法研究大环分子的构象预组织——BCL6抑制剂的案例分析
摘要:本文介绍了构象预组织概念、理论计算方法及其在药物设计中的应用。根据这个理论,人们预期化合物的结合亲和力正比于其构象系综中接近生物活性构象的比例。构象系综中活性构象比例高的化合物比低的化合物具有更高的活性。并以文献报道的BCL6抑制剂8、11为例,演示了如何生成构象系综,并用Flare QM对构象系综进行分级优化、计算活性构象的比例。结果表明,化合物8、11构象系综的活性构象比例与BCL IC50的趋势一致,这为大环化合物的设计提供了一个非常有价值、实用的工具。
肖高铿/2024-05-06
前言
构象预组织(Conformational preorganization)
构象预组织1指的是药物分子在未与生物靶标结合时,已经处于或者能够容易地达到一个构象状态,这个状态与它在结合时的生物活性构象相似或一致。一个预组织的分子可能会在结合时经历较小的构象变化,因此需要的能量(reorganization energy,重组能量)也较小。
重组能量(reorganization energy)
这是指药物分子从其自由状态(未结合状态)转变为与生物靶标结合的状态时所需吸收或释放的能量。这个能量变化涉及到药物分子内部结构的调整,包括原子间距离、键角、二面角的变化,以及内部非共价相互作用的重新分布。重组能量(reorganization energy)1在蛋白-配体结合时也称为构象能惩罚(conformational energy penalty)2或者张力能(strain energy)3。
如果一个药物分子在溶液中已经处于一个接近其生物活性构象的状态(即高度预组织的),那么它在结合过程中所需的重组能量可能较低,因为它不需要经历大幅度的构象变化来适应靶标。相反,如果药物分子在溶液中的自由状态与生物活性构象相差较远,那么它在结合时可能需要较大的重组能量,因为它需要较大的构象调整来适应靶标的结合位点。也就是说,如果一个药物分子在未结合状态下已经接近其生物活性构象,那么它可能更容易与靶标结合,因为所需的内分子重组能量较低。相反,如果药物分子需要经历显著的构象变化才能与靶标结合,那么这可能需要较高的能量成本,从而影响其结合亲和力。
构象预组织用于大环分子设计的案例:BCL6抑制剂8与11
化合物8、11是McCoull等人4发现的BCL6抑制剂。NMR实验表明,在溶液中8是个柔性的化合物,这使得有机会通过构象约束策略来提高活性;而大环化合物11在溶液中预组织为生物活性构象,这使得11的活性较8得以提高。体外实验结果表明,11的IC50=2.9 nM,较8的IC50=370 nM提高了120倍。
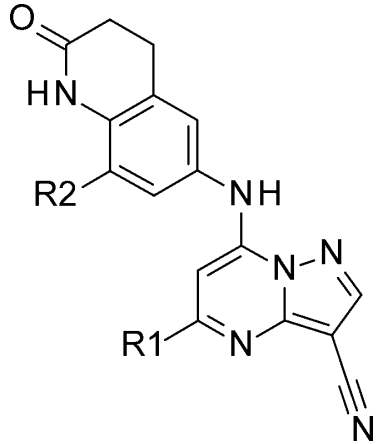
| Compound | R1 | R2 | PDB code | BCL6 FRET IC50 (nM) |
|---|---|---|---|---|
| 8 |  |
H | Model | 370 |
| 11 |  |
5N1Z | 2.9 | |
图1. BCL6抑制剂8与11的化学结构式1
本文的目的
此前,我们已经成功地演示过如何用Spark的大环化工作流实现从线型化合物出发设计大环化合物5-8,在这些案例中,主要应用打分函数对设计的化合物进行排序,但并未从构象预组织角度对设计的大环化合物进行优先级排序。本文以BCL6抑制剂8与11为例,主要目的是演示如何用Flare QM9计算它们在溶液中的构象分布,用构象预组织理论来解释8与11的活性差异,为大环化合物的设计提供非常有价值的、实用的评价工具。
结果
构象系综的产生
8与11在水溶液中的构象系综采用Udvarhelyi等人10提出的ReSCoSS工作流来产生,分三个步骤在三种不同的理论级别方法上计算完成:
- Level_0:MMFF94
- Level_1:R2SCAN-3c/DEF2-mTZVPP//xTB-GFN2
- Level_2:PW6B95-D4/DEF2-TZVPD/IEFPCM-Water
用CDPKit软件包的confgen11,12生成构象,其中用MMFF94力场评估构象能,用RMSD=0.5作为重复构象的阈值,保留构象能\(\le 50\)kcal/mol的构象,得到初始的构象系综:ensemble_level0.sdf。
以Level_0的构象系综为输入,在xTB-GFN2理论水平下进行几何优化,用RMSD=0.125Å作为重复构象的阈值进行构象去重;然后在R2SCAN-3c/DEF2-mTZVPP/IEFPCM-water理论水平下进行单点能计算,得到构象的电子结构能量\(E^{DFT}_{water}\);接着用xTB在GFN2-xTB/alpb-water理论水平计算熵对自由能的贡献(\(G_{mRRHO}\));然后根据SPH方法(见方法部分)将电子结构能量与熵贡献根据式4相加得到构象自由能G,保留\(G\le 9\)kcal/mol的构象,得到新的构象系综:ensemble_level1.sdf。
以Level_1的构象系综为输入,在PW6B95-D4/DEF2-TZVPD/IEFPCM-Water理论水平下进行几何优化,得到构象的电子结构能量\(E^{DFT}_{water}\);接着用xTB在GFN2-xTB/alpb-water理论水平计算熵对自由能的贡献(\(G_{mRRHO}\));然后根据SPH方法(见方法部分)将电子结构能量与熵贡献根据式4相加得到构象自由能G,保留\(G\le3\)kcal/mol的构象,得到最终的构象系综:ensemble_level2.sdf。
上述ReSCoSS工作流除了Level_0之外,QM计算采用pyflare脚本9完成,具体见方法部分。这是一个构象数递减、精度逐步提高的过程,这么做的主要目的是尽可能地减少计算量,而精度损失较小。结果如表1所示,随着计算的进行,系综的构象数越来越少。
表1. 在不同理论级别下计算得到的构象系综的大小
| Method | 8 | 11 |
|---|---|---|
| Level_0 | 222 | 308 |
| Level_1 | 71 | 87 |
| Level_2 | 48 | 8 |
化合物11的构象系综分析
在11的水溶液构象系综中,构象A-E等5个构象的波尔兹曼权重大于1%,总共占据了构象空间的98%以上,如表2所示。用Flare的Conf & align9将这5个构象叠合到从PDB 5N1Z共晶结构提取出来的11生物活性构象上同时计算形状与静电相似性,结果见表2的Similarity列;接着用OpenBabel的obrms工具13计算叠合的结果与生物活性构象之间的重原子isormsd(不包括氢),结果见表2的RMSD列。
表2. 化合物11的构象系综分析
| Conformer | ΔG(kcal/mol) | Boltz. Weight | Similarity | RMSD(Å) |
|---|---|---|---|---|
| A | 0 | 0.668 | 0.901 | 0.39 |
| B | 0.669 | 0.216 | 0.635 | 2.12 |
| C | 1.607 | 0.044 | 0.827 | 0.58 |
| D | 1.614 | 0.044 | 0.834 | 0.51 |
| E | 2.350 | 0.013 | 0.654 | 2.32 |
在构象系综中,构象A、C、D与生物活性构象不仅具有高度3D相似性,这体现在形状与静电场相似打分值Similarity\(\gt\)0.8;此外,这些构象与生物活性构象之间的RMSD\(\lt\)1.0Å,这达到了一般认为的重现生物活性构象的水平。由于这3个构象累积波尔兹曼权重=75.6%,可以认为:在水溶液中,化合物11的75.6%构象空间预组织为生物活性构象。
丰度最高的构象A与生物活性构象相比,如图2(右)所示,原子间距离最大的片段位于内酰胺环的羰基及其α位C原子。其中酰胺α位C原子间的距离最大(d=0.9Å),其它部分的重原子基本完全重合。经过xTB-GFN2与密度泛函几何优化之后,在四氢吡咯环上羟甲基的末端极性氢指向嘧啶并吡唑母核的芳香氮原子,形成分子内氢键,如图2(左)所示。
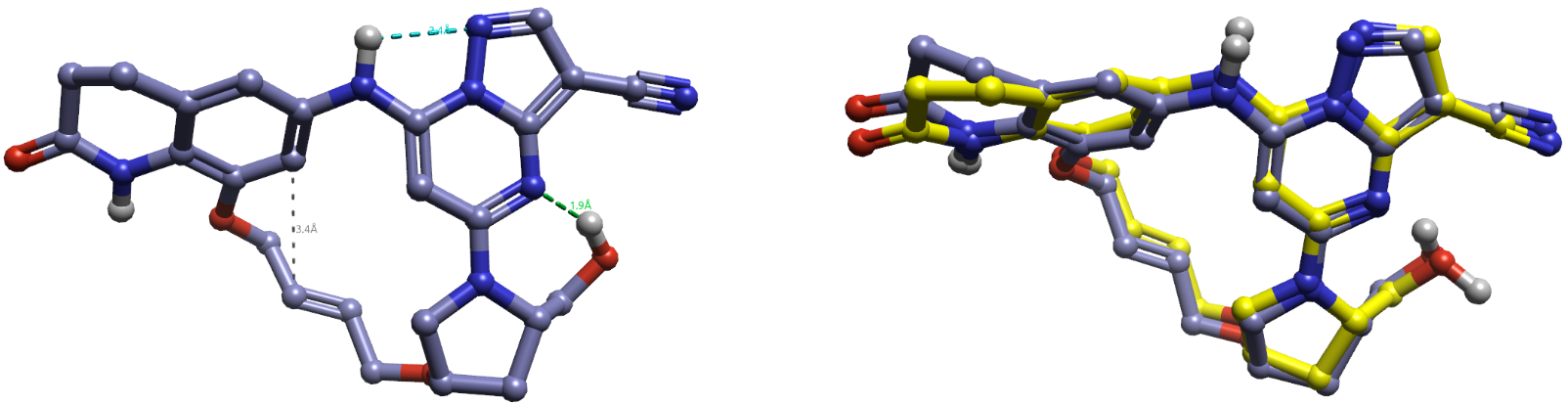
图2. 化合物11的最低能构象A(左)及其与生物活性构象的叠合(右)。黄色:生物活性构象;紫灰色:构象A。
如图3-右所示,构象C、D的主要区别在于内酰胺的一个碳原子翻转,其余原子完全重合,它们的构象能也非常接近。构象C、D与生物活性构象的区别一方面在于内酰胺,另一方面在于四氢吡咯环上羟甲基取向,如图3-左所示。
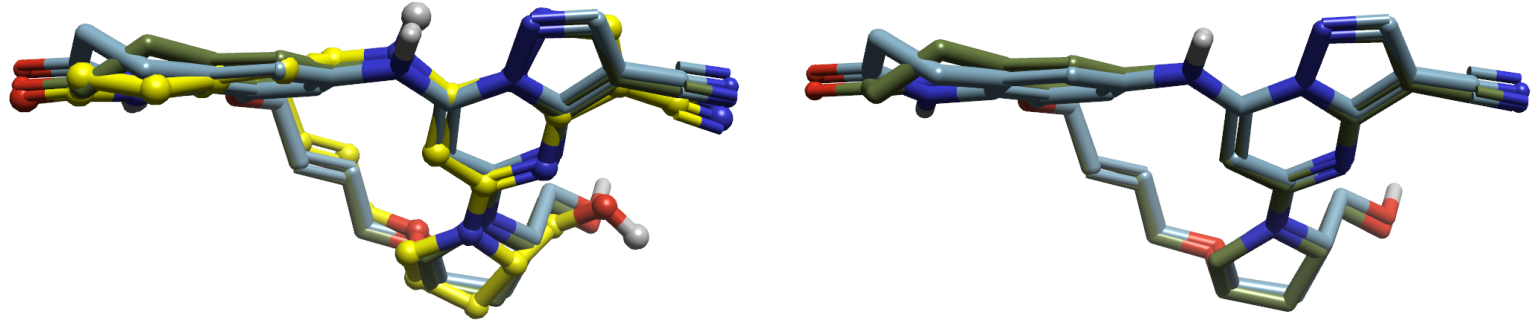
图3. 化合物11的构象C、D与生物活性构象的叠合。黄色:生物活性构象;青灰色:构象C;深绿色:构象D。
构象B、E与生物活性构象相比的3D相似性\(\lt\)0.7,而重原子RMSD\(\gt\)2.0Å,毫无疑问,构象B、E不是药化上所认为的生物活性构象。由于B、E累积波尔兹曼权重=22.9%,因此在11的水溶液中,非活性构象B、E占构象空间的22.9%。
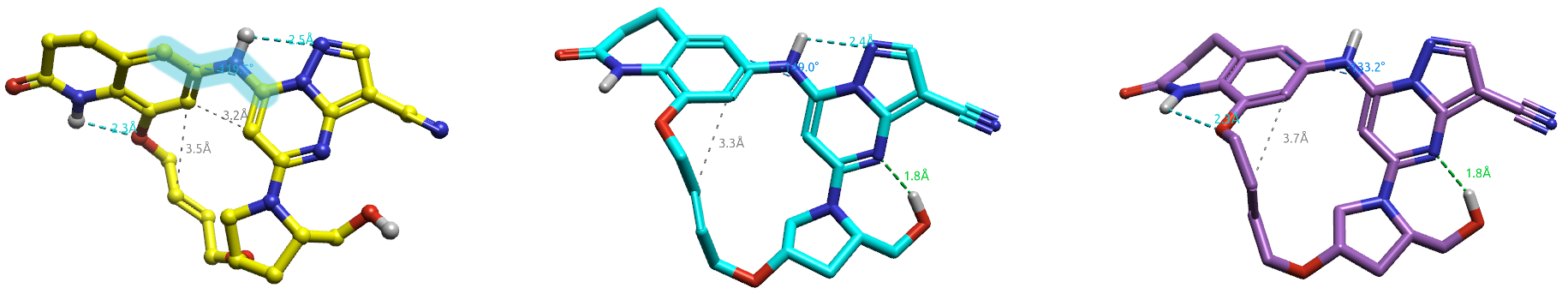
图4. 化合物11的构象B(蓝绿色,中间)、E(紫色,右)与生物活性构象(黄色,左)的比较
在11的生物活性构象中,如图4-左高亮原子所示的两个Ar-NH之间的两面角C-C-N-C=120°。在B、E构象中,这个两面角发生旋转,分别为-149°、-133°。这导致除了嘧啶并吡唑以及四氢吡咯上的羟甲基之外,与生物活性构象比,其它原子都发生了很大的偏移。如下图5所示,在大环连接臂上的原子间距最大,可达6.8Å。
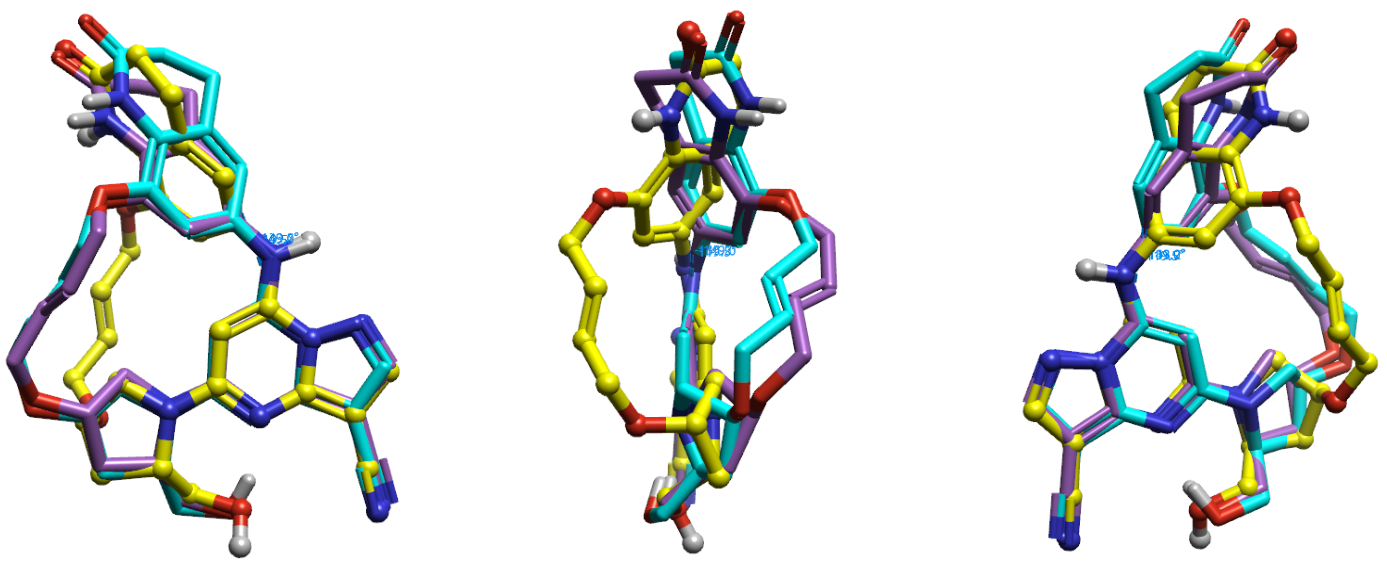
图5. 化合物11的构象B(蓝绿色)、E(紫色)与生物活性构象(黄色)的叠合
化合物8的构象系综分析
化合物8是11的无环化合物,尚未有8与BCL6的共晶结构,它的生物活性构象未知。虽然如此,但可去掉11的大环连接臂来得到8的生物活性构象。这么处理的依据是:无环衍生物7(图6-右,紫色)的生物活性构象(PDB 5N21)与11(图6-中,绿色)的生物活性构象(PDB 5NZ1)的重原子叠合基本一致。
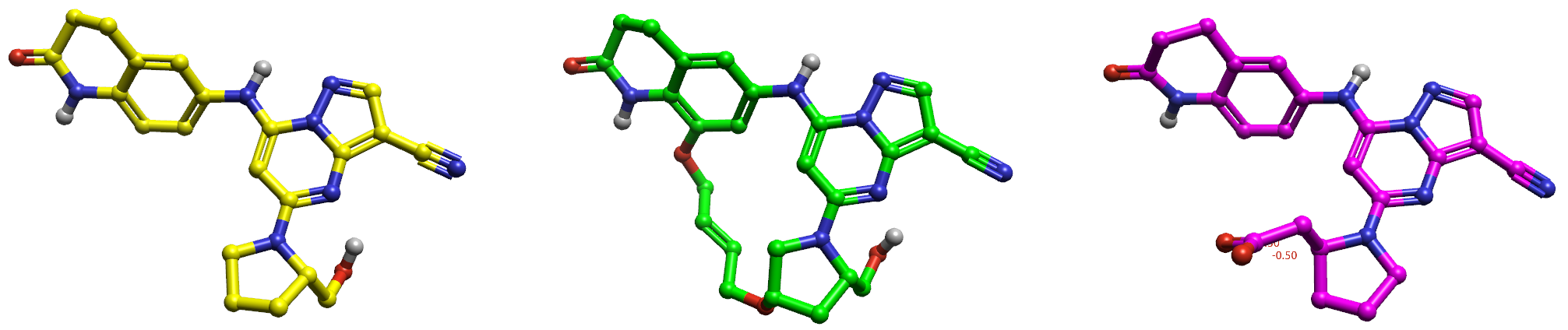
图6. 化合物8的生物活性构象(左,用DFT优化过)、化合物11的生物活性构象(中,用DFT优化过)、化合物7的生物活性构象(右)
在R2SCAN-3c/DEF2-mTZVPP理论水平对8的生物活性构象进行几何优化,与优化前相比,重原子之间的RMSD=0.505,因此可认为8的生物活性构象是一个局部极小点。如图7所示,几何优化过的8与11的生物活性构象相比,除了羟甲基四氢吡咯片段略有差异之外,其它部分的重原子几乎完全重合。其中,羟基氧间的距离最大,d=2.1Å。

图7. 化合物8的生物活性构象几何优化后(黄色)与化合物11的生物活性构象(绿色)
化合物8几何优化后显著的变化之一是phenyl-NH-pyrimidine之间的C-C-N-C两面角τ1(如图8高亮的原子所示),由生物活性构象的120°变化为135~140°。在R2SCAN-3c/DEF2-mTZVPP理论水平对这个两面角进行扭转角分析,可以发现135~140°为优势的低能构象。如前所述,即便该两面角变化20°,但是对重原子的RMSD影响很小。
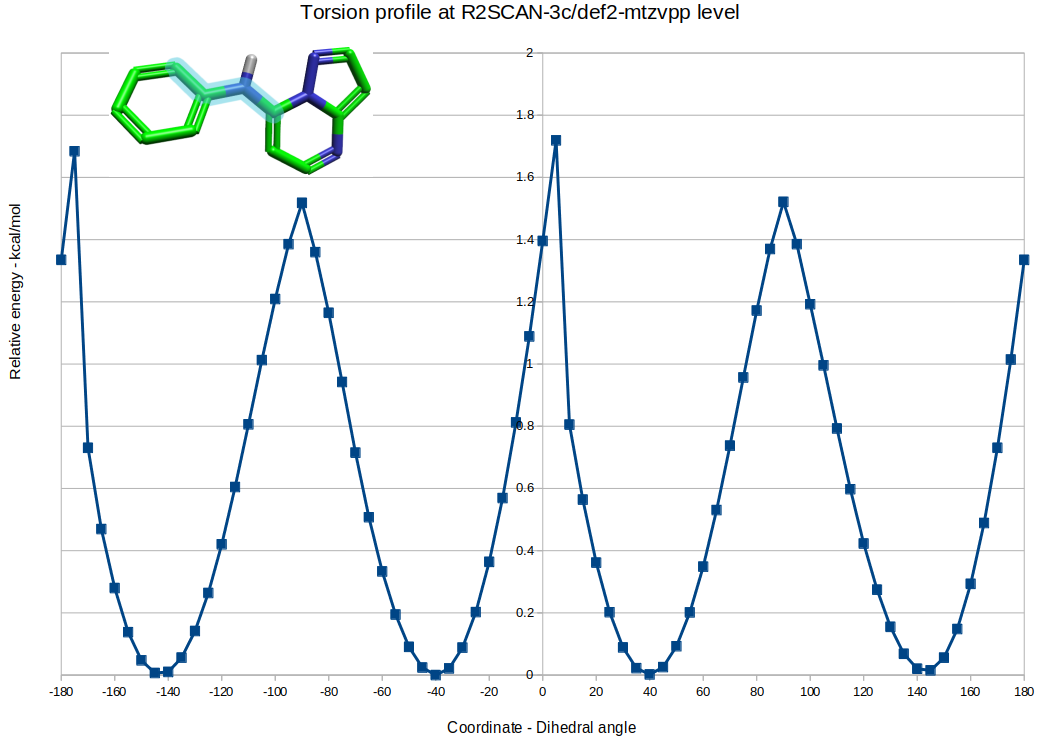
图8. 化合物8的Ar-NH-Ar之间的C-C-N-C两面角τ1的扭转角分析
根据phenyl-NH-pyrimidine之间的C-C-N-C两面角(τ1),化合物8构象系综的构象可以分为两类:活性与非活性。如表3所示。其中活性构象共8个,特征在于τ1在140°左右,而τ2在0°左右;非活性构象共40个,特征在于τ1为-140、-40、40°左右,或者τ1为180°。从波尔兹曼权重上看,在化合物8的构象空间里,预组织为生物活性构象的构象占构象空间的23%,而非活性构象占据构象空间的73%。
表3. 化合物8的构象系综分析
| Conformer(Class/Size) | ΔG(kcal/mol) | Boltz. Weight | Similarity | RMDS(Å) |
|---|---|---|---|---|
| 活性/8 | 0.255~1.931 | 22.78% | 0.902~0.971 | 0.21~0.45 |
| 非活性/40 | 0.0~2.825 | 77.22% | 0.663-0.886 | 0.916~2.260 |
将化合物8的生物活性构象叠合到化合物11的生物活性构象上,如图9所示,重原子与化合物11生物活性构象的几乎重合,RMSD在0.21-0.45Å之间。
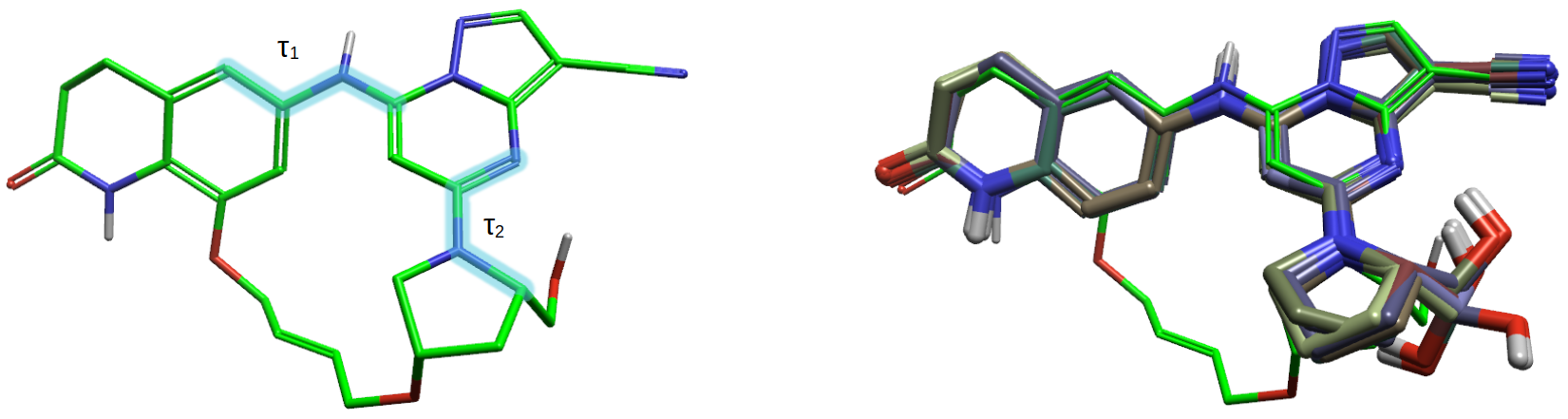
图9. 化合物11的生物活性构象(绿色)与化合物8的8个生物活性构象(棍棒模型)叠合比较。绿色:化合物11(PDB 5NZ1)。
分析40个非活性构象,按照τ1与τ2角度分布,可以分为4类,如表1所示。产生非活性构象的主要原因是:1)τ1发生旋转而不在140°附近;或者2)τ2发生翻转而不在0°附近。
表4. 化合物8非活性构象的τ1与τ2的角度分布
| 类别 | τ1 | τ2 | 构象数 |
|---|---|---|---|
| 1 | -140° | 0/180° | 10 |
| 2 | -40° | 0/180° | 13 |
| 3 | 40° | 0/180° | 14 |
| 4 | 140° | 180° | 3 |
将非活性构象叠合到化合物11上,如图10所示,可以观察到1-3类是因为τ1旋转而使得内酰胺环取向变化,这导致内酰胺环不能与结合位点继续发生极性相互作用,同时也可能傍随着τ2的翻转。而4类构象虽然内酰胺部分为生物活性构象,但是因为羟甲基四氢吡咯片段的旋转,也就是τ2翻转而导致这些构象为非活性。
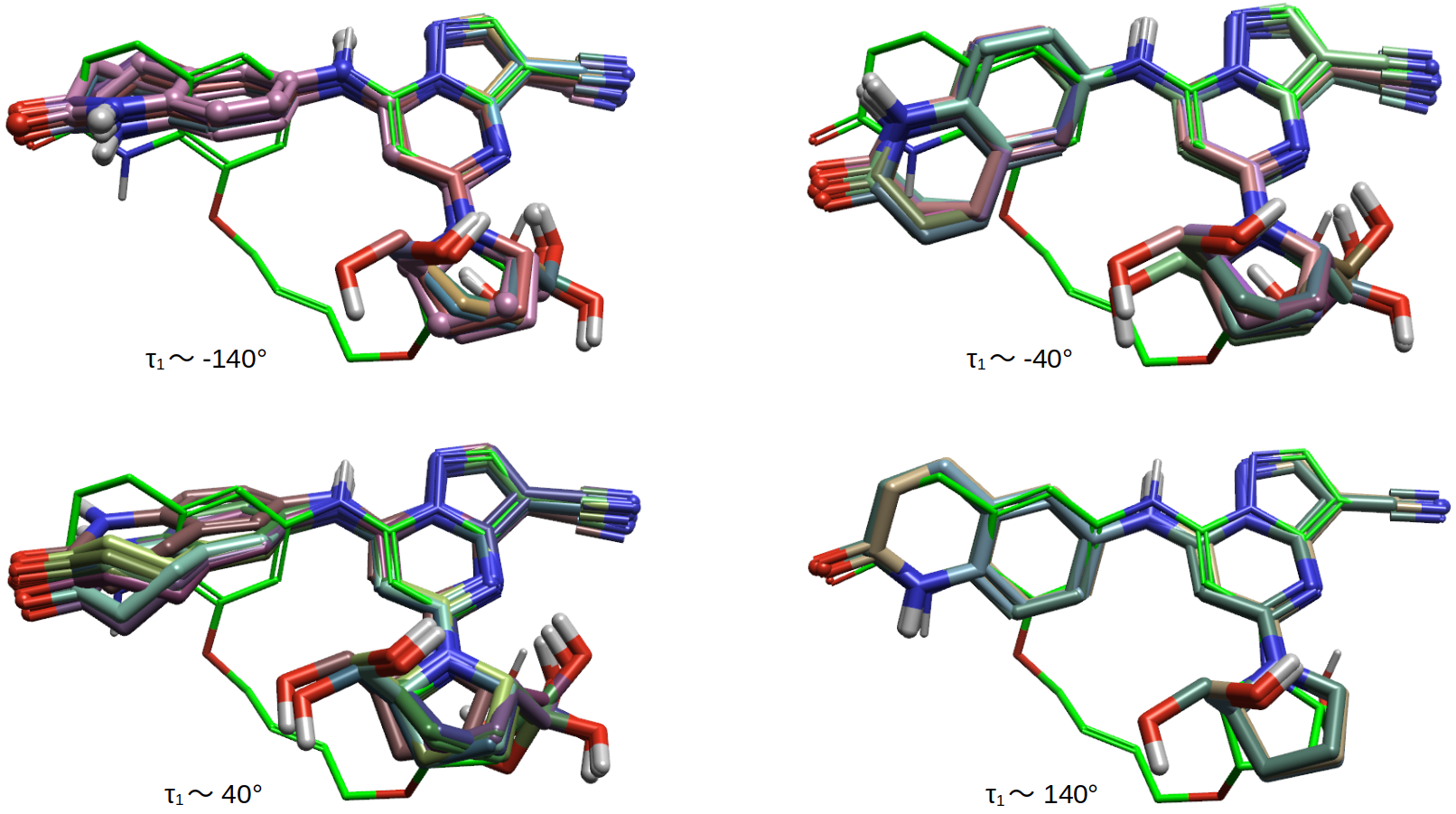
图10. 化合物8的40个非生物活性构象(棍棒模型)与化合物11的生物活性构象(绿色)比较
构象系综与结合亲和力的关系
人们预期化合物的结合亲和力正比于其构象系综中接近生物活性构象的比例。有了构象系综,多种方式可以用来评估生物活性构象与亲和力的关系。在McCoull等人4的研究中,如果一个构象与实验构象之间的RMSD\(\lt\)0.5Å,则定义为活性构象,然后计算活性构象数比例并与结合亲和力进行比较分析。而Sindhikara等人14则在McCoull方法的基础上融合了波尔兹曼权重,比较线型化合物与大环衍生物的公共子结构,并计算公共子结构部分的RMSD,然后计算构象系综的波尔兹曼加权平均RMSD,记为\(\langle RMSD_{cons}\rangle\),然后将\(\langle RMSD_{cons}\rangle\)与结合亲合力进行比较。而OpenEye的FreeForm15,16则直接计算结合构象的重组构象自由能,这个方法的好处是简单且直接。
在McCoull等人4的研究中,测试了5个BCL-6抑制剂,它们的构象系综活性构象比例与生物活性(BCL6的IC50)之间的相关性系数R2=0.86,并成功的预测了新化合物的活性趋势。由于大环化合物的合成成本很高,因此用这个方法在合成之前预测线型化合物大环化之后是否活性得以保持或改善具有非常重要的意义。
在本文中,用RMSD与关键两面角一起将构象系综划分为活性与非活性构象,然后统计活性构象的累积波尔兹曼权重并作为活性构象的比例。在线型化合物8、大环化合物11的构象系综中活性构象的权重分别为22.8%、75.6%,这与它们的生物活性趋势是一致的,BCL6 IC50分别为370、2.9 nM。
表5. 化合物8、11的构象分布与结合自由能
| Compound | Proportion of bio-conformer | ΔGbinding (kcal/mol) |
|---|---|---|
| 8 | 22.6% | -8.8 |
| 11 | 75.6% | -11.6 |
方法与材料
结构准备
将11与BCL6的共晶结构PDB 5N1Z下载到Flare1中,并用Protein Prep工具进行结构准备以添加氢原子、优化氢键、消除原子冲突并给蛋白结构与配体分配最佳质子化状态,任何截短的蛋白质链被封端作为蛋白质准备的一部分。将配体提取出来,导出作为生物活性构象(11_bound.sdf)。将导出的配体复制为smiles格式(11.smi),粘贴到Flare的配体表单,然后用“Pop to 3D”生成一个计算3D结构,并保存为11.sdf,其作为构象搜索的起点。
化合物8是从11开始,在Flare里用结构编辑器删除大环连接臂得到8的结合模式(8_bound.sdf)。将导出的配体复制为smiles格式(8.smi),粘贴到Flare的配体表单,然后用“Pop to 3D”生成一个计算3D结构,并保存为8.sdf,其作为构象搜索的起点。
化合物7与BCL6的共晶键结构PDB 5N21用Flare进行同样的处理,得到7的生物活性构象。
用CDPKit进行构象搜索
CDPKit的confgen不仅适合于类药分子,也适合于大环化合物2,3。以11为例,用confgen生成构象系综:
1 2 3 4 | confgen -C LARGE_SET_DENSE \
-e 50 \
-n 2000 \
-i 11.sdf -o 11-ensemble.sdf |
在GFN2-xTB理论水平进行几何优化
用pyflare的qm.py脚本来调用xTB17,18进行计算,以上一步的11-ensemble.sdf为例:
1 2 | pyflare qm.py --method GFN2-xTB -T VeryTight \ -I 500 --max-threads 24 -M 24 11-ensemble.sdf >> 11-ensemble-xtb.sdf |
在DFT理论水平进行QM计算
所有的DFT理论水平计算采用Flare QM内置PSI419,20完成,也可以通过pyflare的qm.py脚本完成,或者直接使用PSI4。
波尔兹曼权重计算
构象系综中每个构象的波尔兹曼权重根据构象的自由能(G)计算,取相对值(ΔG)时,通常将能量最低的构象定为零点,根据平衡常数与自由能变的关系可以得到:
$$
q = e^{-ΔG \over {RT}}\cdots (1)
$$
其中R为气体常数,T为温度。构象i出现的概率(Pi)服从玻尔兹曼分布:
$$
P_i = {q_i \over \sum{q_i}}\cdots (2)
$$
构象自由能G
构象自由能G是根据Spicher等人21的SPH(Single Point Hessian)方法计算:
$$
G = {E^{DFT}_{water} + G^{GFN2-xTB/alpb-water}_{mRRHO}}\cdots (3)
$$
比如在Level_2时,\(E^{DFT}_{water}\)是在PW6B95-D4/DEF2-TZVPD/IEFPCM-Water理论水平计算的电子结构能量,其中包含了溶剂效应。\(G_{mRRHO}\)是用xTB在GFN2-xTB/alpb-water理论水平对DFT的几何优化结果计算得到,已经包含了溶剂效应。
结论
本文介绍了构象预组织概念以及理论计算方法,并介绍了其在药物设计中的应用。根据这个理论,人们预期化合物的结合亲和力正比于其构象系综中接近生物活性构象的比例。构象系综中活性构象比例高的化合物比低的化合物具有更高的活性。并以BCL6抑制剂8、11为例,演示了如何生成构象系综,并用Flare QM对构象系综进行分级优化、并计算活性构象的比例。结果表明,化合物8、11构象系综的活性构象比例与BCL IC50的趋势一致,这为大环化合物的设计提供了一个非常有价值、实用的工具。
文献
- Foloppe, N. and Chen, I.J. (2016) “Towards understanding the unbound state of drug compounds: Implications for the intramolecular reorganization energy upon binding,” Bioorganic and Medicinal Chemistry, 24(10), pp. 2159–2189. Available at: https://doi.org/10.1016/j.bmc.2016.03.022.
- Boström, J., Norrby, P.O. and Liljefors, T. (1998) “Conformational energy penalties of protein-bound ligands.,” Journal of computer-aided molecular design, 12(4), pp. 383–96. Available at: https://doi.org/10.1023/a:1008007507641.
- Perola, E. and Charifson, P.S. (2004) “Conformational Analysis of Drug-Like Molecules Bound to Proteins: An Extensive Study of Ligand Reorganization upon Binding,” Journal of Medicinal Chemistry, 47(10), pp. 2499–2510. Available at: https://doi.org/10.1021/jm030563w.
- McCoull, W. et al. (2017) “Discovery of Pyrazolo[1,5-a]pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors,” Journal of Medicinal Chemistry, 60(10), pp. 4386–4402. Available at: https://doi.org/10.1021/acs.jmedchem.7b00359.
- Matthias Baue. 用SPARK设计大环BRD4抑制剂. 墨灵格的博客. 2018-02-22. http://blog.molcalx.com.cn/2018/03/09/using-spark-to-design-macrocycle-brd4-inhibitors.html
- 肖高铿. 文献重现 | 大环EGFR激酶抑制剂BI-4020的先导化合物5与6的发现与设计. 墨灵格的博客. 2020-01-18. http://blog.molcalx.com.cn/2020/01/18/egfr-bi-4020.html
- 肖高铿. 用Spark重现大环HPK1抑制剂的设计. 墨灵格的博客. 2023-02-23. http://blog.molcalx.com.cn/2023/02/23/using-spark-to-design-macrocycle-hpk1-inhibitors.html
- 肖高铿. 用SPARK重现选择性大环CDK2抑制剂QR-6401的设计. 墨灵格的博客. 2023-02-26. http://blog.molcalx.com.cn/2023/02/26/using-spark-to-design-macrocycle-cdk2-inhibitors.html
- Flare V8.0. http://www.cresset-group.com/software/flare
- Udvarhelyi, A., Rodde, S. and Wilcken, R. (2021) “ReSCoSS: a flexible quantum chemistry workflow identifying relevant solution conformers of drug-like molecules,” Journal of Computer-Aided Molecular Design, 35(4), pp. 399–415. Available at: https://doi.org/10.1007/s10822-020-00337-7.
- CDPKit Version 1.1.0. https://github.com/molinfo-vienna/CDPKit
- Seidel, T. et al. (2023) “High-Quality Conformer Generation with CONFORGE: Algorithm and Performance Assessment,” Journal of Chemical Information and Modeling, 63(17), pp. 5549–5570. Available at: https://doi.org/10.1021/acs.jcim.3c00563.
- OpenBabel Version 3.1.0. Open Babel – the chemistry toolbox. https://openbabel.org
- Sindhikara, D. and Borrelli, K. (2018) “High throughput evaluation of macrocyclization strategies for conformer stabilization,” Scientific Reports, (April), pp. 1–8. Available at: https://doi.org/10.1038/s41598-018-24766-5.
- FreeForm. https://docs.eyesopen.com/applications/szybki/freeform/freeform.html
- 肖高铿. FreeForm–构象稳定性评估及其在先导化合物优化中的应用. 墨灵格的博客. 2016-07-02. http://blog.molcalx.com.cn/2016/07/02/freeform-conformer-stability.html
- xTB Version 6.7.0. Semiempirical Extended Tight-Binding Program Package. https://github.com/grimme-lab/xtb
- Bannwarth, C. et al. (2021) “Extended tight-binding quantum chemistry methods,” Wiley Interdisciplinary Reviews: Computational Molecular Science. Blackwell Publishing Inc. Available at: https://doi.org/10.1002/wcms.1493.
- https://github.com/psi4/psi4
- Smith, D.G.A. et al. (2020) “PSI4 1.4: Open-source software for high-throughput quantum chemistry,” Journal of Chemical Physics, 152(18). Available at: https://doi.org/10.1063/5.0006002.
- Spicher, S. and Grimme, S. (2021) “Single-Point Hessian Calculations for Improved Vibrational Frequencies and Rigid-Rotor-Harmonic-Oscillator Thermodynamics,” Journal of Chemical Theory and Computation, 17(3), pp. 1701–1714. Available at: https://doi.org/10.1021/acs.jctc.0c01306.
联系我们、获取试用
想要亲自计算或在自己的项目中使用Flare QM,或者商务合作,请联系我们。


