
警惕虚拟筛选性能测试的结论——选择合理的蛋白结构来提高富集性能
摘要:本文以DUD-E的靶标HIV PR为例,演示了蛋白结构选择对虚拟筛选性能的影响。具体的讲,当使用PDB 1XL2作为分子对接的蛋白结构时,Flare docking与文献报道的Glide、GOLD等方法的富集性能差不多,都没有显著地优于空模型;当使用PDB 1RPO作为分子对接的蛋白结构时,Flare docking表现出优秀的富集性能。这说明,用分子对接方法对靶标进行虚拟筛选时,选择不同的蛋白结构可导致截然相反的性能评估结论。这警示我们:文献报道的虚拟筛选性能评测差的方法不一定真的差,难以虚拟筛选的靶标可能不是真的难,也许只是蛋白结构选择的问题。
肖高铿/2023-09-01
前言
人们普遍接受用公开的基准数据集对分子对接等虚拟筛选方法进行性能测试,并用于计算方法选择与推荐。有的靶标,因为在测试中各种软件的性能均表现不佳,被认为是难以用分子对接成功虚拟筛选的靶标。在DUD-E基准测试集中的HIV PR1,2就是一个这样的靶标。根据最近的文献报道3,4,在该靶标上,流行的分子对接软件均没有在早期富集性能上优于不用任何计算的空模型(Null model)。如下表1所示,GLIDE与GOLD的性能表现并没有优于分子量与氢键供体数代表的空模型。
表1. 文献报道的分子对接虚拟筛选方法在DUD-E HIVPR上的性能指标
| Metric | Glide SP* | GOLD ChemPLP* | MolWt | NumHDonors |
|---|---|---|---|---|
| AUC | 0.715 | 0.392 | 0.750 | 0.676 |
| logAUCλ=0.1% | 30.3 | 11.0 | 46.9 | 31.5 |
| adj-logAUCλ=0.1% | 15.8 | -3.5 | 32.4 | 17.0 |
| BEDROCα=321.9 | 0.171 | 0.053 | 0.342 | 0.168 |
| BEDROCα=80.5 | 0.152 | 0.040 | 0.430 | 0.156 |
| BEDROCα=20.0 | 0.242 | 0.048 | 0.532 | 0.230 |
| EF0.5% | 10.0 | 2.1 | 27.9 | 13.74 |
| EF1% | 8.6 | 2.1 | 26.3 | 11.7 |
| EF5% | 4.6 | 0.9 | 11.5 | 6.27 |
| EF10% | 3.6 | 0.5 | 6.1 | 3.1 |
*Glide与GOLD的性能指标是根据Shen等人4报道的打分值重新统计得到。
Chaput等人4在DUD-E数据集上测试了Glide、GOLD、Surflex与FlexX等四种分子对接方法的虚拟筛选性能,将BEDROCα=80.5不小于0.5做为虚拟筛选成功的标准。结果表明,这4个分子对接方法在HIV PR这个靶标上的BEDROCα=80.5分别为0.14、0.3、0.1与0.05,4个虚拟筛选方法不仅未达到成功的标准,而且更重要的是没有优于空模型(MolWt,BEDROCα=80.5=0.43)。
然而,成功的分子对接虚拟筛选取决于多个方面,在其它条件保持不变情况下,蛋白结构的选择与准备是另一个决定性的因素。如图1所示,HIV PR的分子对接虚拟筛选不仅是个re-docking的过程,更是个cross-docking的过程。可以想象:如果以DUD-E测试集用的PDB 1XL2为蛋白结构,可能除了绿色的配体发生re-docking可以被对接命中之外,其它方形、三角形与正方形的三个配体均不能被命中;如果以PDB 1PRO为蛋白结构进行对接虚拟筛选,除了三角形的分子不能被命中,其它三个可能均可被命中。
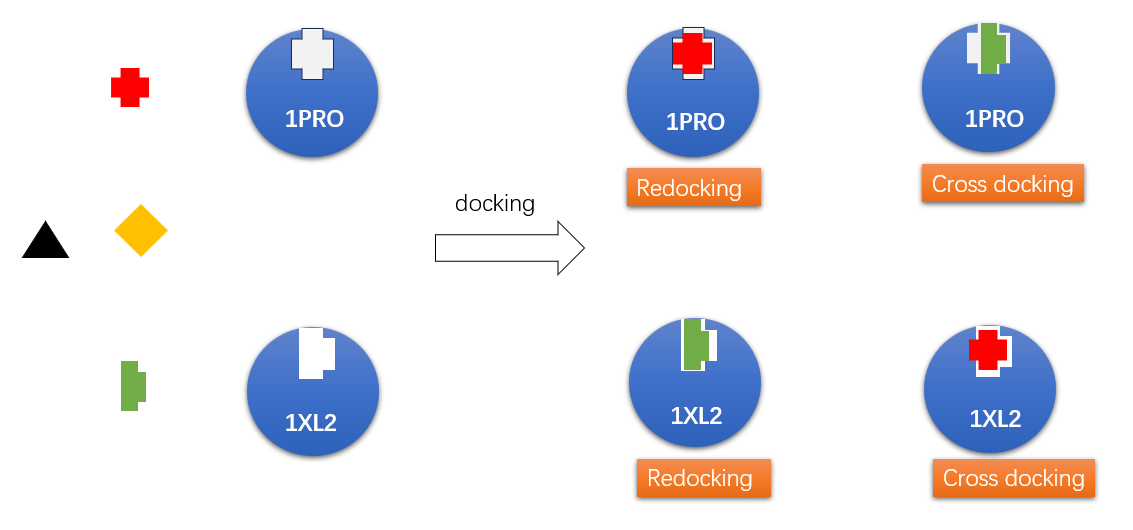
图1. 分子对接虚拟筛选示意图
因此本文的主要目的之一是,以DUD-E的HIV PR靶标为例,演示蛋白选择对Flare Docking的虚拟筛选性能影响。具体的讲,当选择PDB 1PRO作为蛋白结构时,Flare docking表现出优秀的虚拟筛选性能;而选择PDB 1XL2作为蛋白结构时,仅表现出与空模型想当的性能。
本文的另一个目的是让大家警惕虚拟筛选性能测试的结论,提醒基准测试的目的:仅仅评估在当前使用条件下虚拟筛选方法的性能。它不能评估一个软件对该靶标是否适用,有可能DUD-E指定的那个蛋白结构并不适用于你正在测试的方法。分子对接对蛋白结合口袋非常敏感,有可能只是数据集中的active分子大部分不能与你选择的蛋白结合口袋3D互补,而不是软件的性能差。
方法
分子对接虚拟筛选流程
Flare Docking的分子对接计算引擎是Lead Finder5,通过pyflare的docking.py脚本进行分子对接计算,包括包括4个步骤:1)蛋白模型构建;2)能量格点计算;3)在能量格点里对接分子;4)打分。
为了兼顾速度,在本文中Flare docking采用虚拟筛选模式。Lead Finder的打分函数是一种半经验的力场方法,并显式地考虑了不同类型的分子间相互作用。对每个打分成分用一个经验系数进行缩放,得到三种不同目的的打分函数:
- dG-score:预测结合亲和力,在本文中也表示为LF_dG
- VS-score:在虚拟筛选中对不同的分子排序,在本文中也表示为LF_VSscore
- Rank-Score:对结合模式进行排序,在本文中也表示为LF_Rank_Score
蛋白的准备
用Flare下载共晶结构PDB 1XL2与1PRO,并用Protein Prep工具进行结构准备,仔细检查结构以确保配体结构正确、相互作用合理,删除全部的水,仅保留A、B链的蛋白部分与配体,分别导出蛋白结构与配体结构备用。
数据库分子的准备
DUD-E HIV PR数据集2里预先准备好的SDF格式actives(actives_final.sdf)与decoys(decoys_final.sdf)结构直接用于虚拟筛选,不进行额外的准备。
分子对接虚拟筛选
分子对接虚拟筛选使用pyflare的docking脚本,在命令行下计算能量格点并执行虚拟筛选。以PDB 1PRO蛋白结构为例,演示如下。
以共晶配体为参比分子,跑一个redocking,同时进行能量格点计算:
1 2 3 4 5 6 7 8 | pyflare docking.py -p 1pro_prot.pdb -j pdb --protein-chain-types p \ --reference 1pro_ligand.sdf -i sdf \ --write-grid 1pro_prot.grid \ --csv redock.csv \ --max-poses 20 \ --batch-size 120 \ --quality virtual-screening \ 1pro_ligand.sdf -i sdf >> redock.sdf |
计算完毕,生成了一个新的文件1pro_prot.grid即为所需的能量格点文件。
actives虚拟筛选:
1 2 3 4 5 6 7 | pyflare docking.py -p 1pro_prot.pdb -j pdb --protein-chain-types p \ --grid-from-file 1pro_prot.grid \ --csv actives_dock.csv \ --max-poses 20 \ --batch-size 120 \ --quality virtual-screening \ actives_final.sdf -i sdf >> actives_dock.sdf |
计算完毕得到的actives_dock.sdf文件即为actives数据集对接结果文件。
decoys虚拟筛选:
1 2 3 4 5 6 7 | pyflare docking.py -p 1pro_prot.pdb -j pdb --protein-chain-types p \ --grid-from-file 1pro_prot.grid \ --csv decoys_dock.csv \ --max-poses 20 \ --batch-size 120 \ --quality virtual-screening \ decoys_final.sdf -i sdf >> decoys_dock.sdf |
计算完毕得到的decoys_dock.sdf文件即为decoys数据集对接结果文件。
用GNINA进行重新打分
GNINA是一款采用深度学习CNN模型的分子对接软件6,7,在本文中用来对Flare Docking结果再打分。用GNINA对actives对接结果进行重新打分:
1 | gnina --config dock.conf -l actives_dock.sdf -o actives.sdf --score_only |
用GNINA对decoys对结果结果进行重新打分:
1 | gnina --config dock.conf -l decoys_dock.sdf -o decoys.sdf --score_only |
其中dock.conf为gnina对接参数文件,内容如下:
1 2 3 4 5 6 7 | receptor = 1pro_prot.pdbqt center_x = -8.919 center_y = 15.697 center_z = 27.716 size_x = 30 size_y = 30 size_z = 30 |
GNINA打分之后,在sdf文件里包含了3个新的打分值:
- CNNscore: GNINA的CNN模型pose打分值
- CNNaffinity:GNINA的CNN模型结合亲和力预测值
- CNN_VS:CNNscore x CNNaffinity
数据融合
Flare docking对接计算产生LF_VSscore,LF_dG与LF_Rank_Score等多个打分值,其中LF_VSscore用于对化合物进行打分排序。用GNINA重新打分之后,得到三个新的打分值CNNscore,CNNaffinity与CNN_VS。其中CNNscore是pose打分,CNNaffinity是亲和力打分,CNN_VS是CNNscore与CNNaffinity的乘积,即:
CNN_VS = CNNaffinity x CNNscore
在本文中,将CNNscore与LF_VSscore进行相乘:
LF_CNN_VS = LF_VSscore x CNNscore
CNNscore可以与多种打分进行数据融合用于虚拟筛选,我们已经在DUE-E上进行大规模的验证,可以提高大部分靶标的虚拟筛选富集能力。
数据处理与虚拟筛选性能评估
鉴于从DUD-E下载的actives与decoys数据集进行了互变异构体与质子化状态枚举处理,导致一个化合物会以不同的形式多次出现、并可能被虚拟筛选多次命中,因此对虚拟筛选结果按化合物名称进行去重,同名化合物仅以其中打分最高那个来代表,所有的性能评估指标都在去重的基础上进行计算。
在虚拟筛选过程中,有可能有的化合物处理失败而没有出现在结果文件里,为了保证性能评估的结果与其它文献报道的具有可比性,将缺失的化合物人为地分配一个打分值0然后再进行性能评估。
用AUC、logAUC、三种α(321.9,80.5与20.0)值的BEDROC、富集因子(top 0.5%,1%,5%与10%)等指标来评估虚拟筛选的性能,具体的计算方法参见前文10。
结果与讨论
数据处理
对接结果用python pandas聚合函数groupby对化合物分值进行处理:同一个化合物的互变异构体、不同质子化状态仅保留打分最优的那个作为该化合物的打分值。actives与decoys数据的处理结果如表2所示。由于预先准备的结构有部分错误以及对接失败的原因,并不是所有的输入分子都出现在了结果里面。这些处理失败的分子给予一个打分值0之后参与统计,以便不同方法得到的性能指标具有可比性。
表2、DUD-E HIV PR数据集化合物统计
| EGFR数据集 | 输入的化合物数 | 输入的异构体数 | 输出的化合物数 |
|---|---|---|---|
| actives | 536 | 1395 | 523 |
| decoys | 35688 | 36278 | 33500 |
数据融合后的打分结果见附件all_score.csv,去重后用于统计处理的打分结果文件见附件all_score_grouped.csv。
当使用PDB 1XL2作为蛋白结构时的虚拟筛选富集性能没有优于空模型
表3. 当DUD-E HIV PR数据集使用PDB 1XL2时的主要性能指标
| Metric | LF_VSscore | LF_CNN_VS | CNNaffinity | CNN_VS |
|---|---|---|---|---|
| AUC | 0.756 | 0.723 | 0.762 | 0.667 |
| logAUCλ=0.1% | 34.6 | 27.3 | 36.2 | 23.7 |
| adj-logAUCλ=0.1% | 20.1 | 12.9 | 21.8 | 9.3 |
| BEDROCα=321.9 | 0.169 | 0.058 | 0.245 | 0.029 |
| BEDROCα=80.5 | 0.169 | 0.085 | 0.225 | 0.060 |
| BEDROCα=20.0 | 0.306 | 0.186 | 0.325 | 0.151 |
| EF0.5% | 10.4 | 3.3 | 16.0 | 2.0 |
| EF1% | 9.5 | 4.5 | 13.4 | 2.6 |
| EF5% | 6.2 | 3.4 | 6.5 | 2.8 |
| EF10% | 5.0 | 3.1 | 4.6 | 2.5 |
当使用PDB 1XL2作为蛋白结构进行虚拟筛选时的主要性能指标如表3所示。可以看出,与Glide SP(见表1)等方法一样,LF VSscore、LF_CNN_VS、CNNaffinity与CNN_VS也没有表现出比空模型更好的富集性能。一方面,Glide、LF VSscore、LF_CNN_VS、CNNaffinity与CNN_VS等的富集指标EF1% 分别为8.6、9.5、4.5、13.4与2.6,远低于空模型(MolWt)的26.3,如图2所示。标准的对接方法Glide与Flare docking表现出相似的富集性能,深度学习打分函数CNNaffinity虽然提高了50%左右,但也没有表现出比空模型MolWt更好的性能,数据融合打分函数LF_CNN_VS与CNN_VS的富集性能甚至比独立打分函数性能更差。
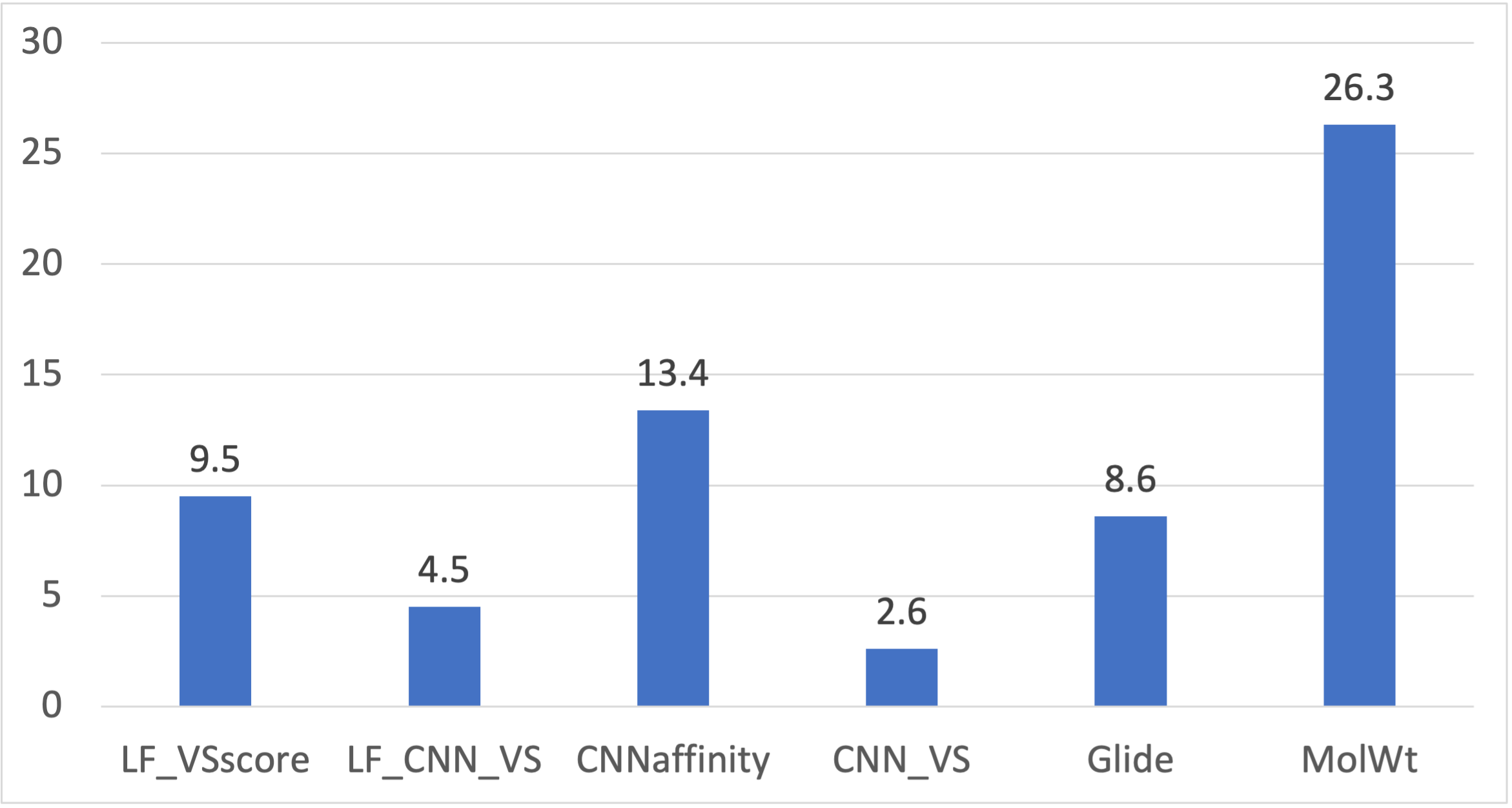
图2. 使用PDB 1XL2作为蛋白结构进行对接虚拟筛选的早期富集性能指标EF1%比较
另一方面,Glide、LF VSscore、LF_CNN_VS、CNNaffinity与CNN_VS等的富集指标BEDROCα=80.5分别为0.152、0.16、0.085、0.225与0.06,远低于空模型(MolWt)的0.43,如图3所示。再一次,标准的对接方法Glide与Flare docking表现出相似的富集性能【深度学习打分函数CNNaffinity大幅地提高了富集性能,但也没有表现出比空模型MolWt更好的性能;数据融合打分函数LF_CNN_VS与CNN_VS的富集性能比独立打分函数性能更差。
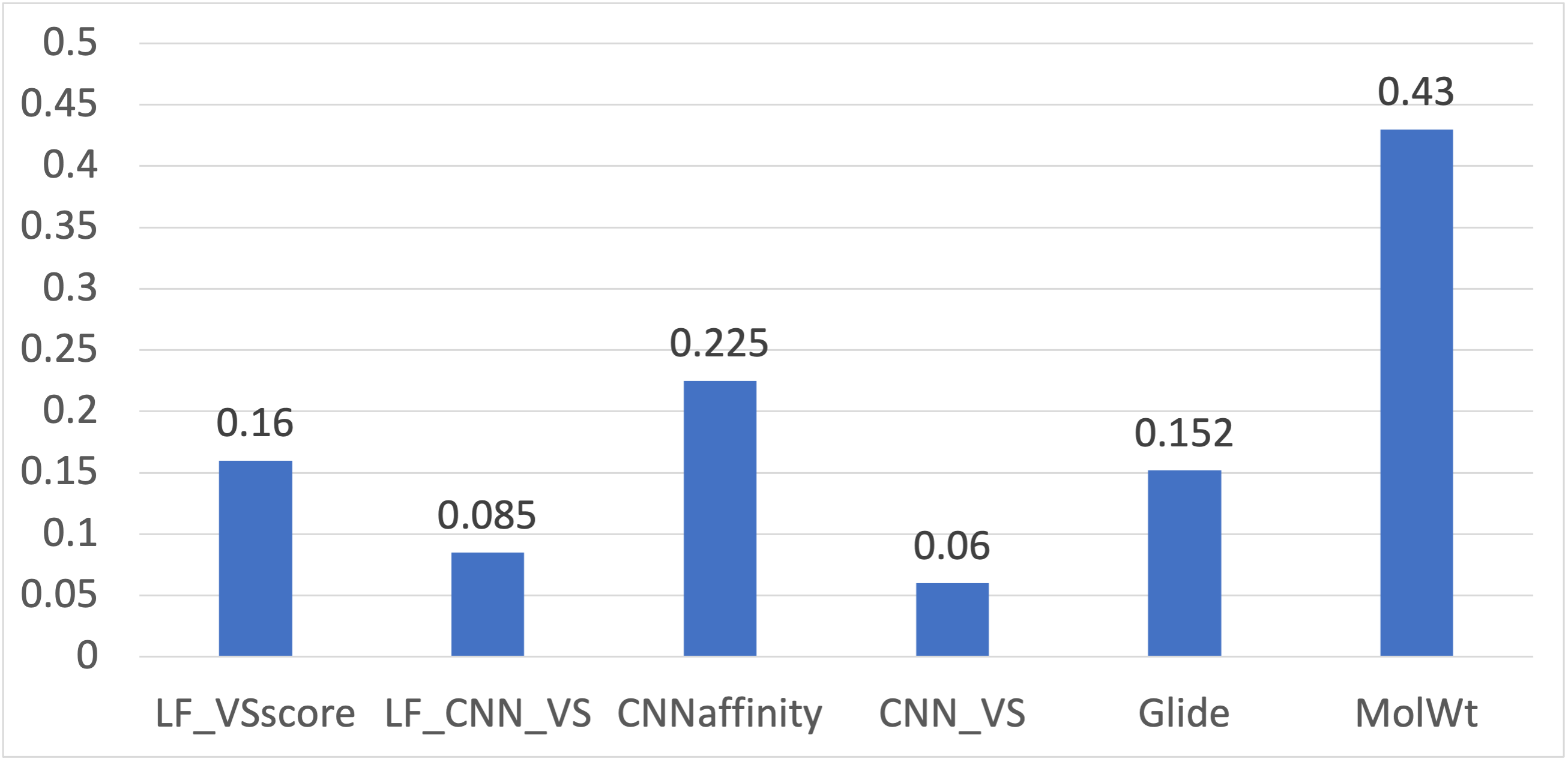
图3. 使用PDB 1XL2作为蛋白结构进行对接虚拟筛选的早期富集性能指标BEDROCα=80.5比较
如果性能测试就到此为止,而不去做更多的计算实验,那么我的一个初步结论将是:HIV PR确实是一个很难的靶标,Flare Docking与其它的分子对接软软件一样,都没有获得比空模型更好的富集性能。
之前我们已经分享过很多的ensemble docking或ensemble vs的研究,相关内容可以从之前的博客里了解到。一个初步的结论是:无论是分子对接还是药效团虚拟筛选,使用越多的蛋白-配体信息,虚拟筛选性能越好。这提醒我们:也许在DUD-E的HIV PR数据集里提供的蛋白结构不适于容纳大部分的actives分子,需要使用更多的蛋白结合口袋形状来进行虚拟筛选。出于这个假设,我们开始了ensemble docking测试,结果发现了PDB 1PRO更适合于Flare docking对这个数据集进行虚拟筛选。
当使用PDB 1PRO作为蛋白结构时的虚拟筛选性能得到大幅提升
表4. 当DUD-E HIV PR数据集使用PDB 1PRO时的主要性能指标
| Metric | LF_VSscore | LF_CNN_VS | CNNaffinity | CNN_VS |
|---|---|---|---|---|
| AUC | 0.890 | 0.918 | 0.910 | 0.910 |
| logAUCλ=0.1% | 54.3 | 61.2 | 66.9 | 57.8 |
| adj-logAUCλ=0.1% | 39.9 | 46.7 | 52.5 | 43.4 |
| BEDROCα=321.9 | 0.350 | 0.598 | 0.714 | 0.509 |
| BEDROCα=80.5 | 0.404 | 0.507 | 0.611 | 0.460 |
| BEDROCα=20.0 | 0.571 | 0.618 | 0.687 | 0.583 |
| EF0.5% | 35.3 | 56.2 | 72.2 | 47.2 |
| EF1% | 27.6 | 39.0 | 48.4 | 35.4 |
| EF5% | 12.7 | 13.1 | 14.2 | 12.1 |
| EF10% | 7.4 | 7.7 | 8.0 | 7.5 |
当使用PDB 1PRO作为蛋白结构时的虚拟筛选性能主要指标如表4所示,显而易见地,LF_VSscore、LF_CNN_VS、CNNaffinity与CNN_VS等打分函数都表现出大幅的性能提升,这一点还可以从图4的ROC曲线直观地看到:使用PDB 1XL2的曲线在最下方,使用PDB 1PRO的曲线在上方。
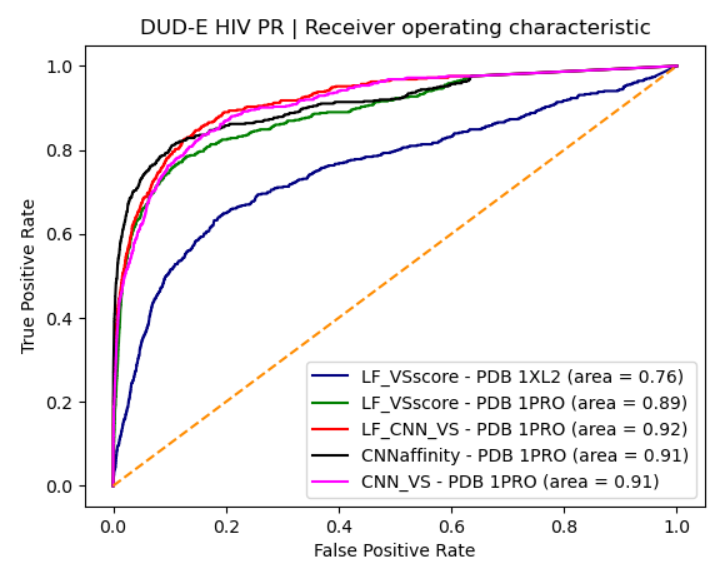
图4. 选用两个不同蛋白结构(PDB 1XL2与1PRO)时的ROC曲线
就LF_VSscore而言,使用新的蛋白结构之后,综合性能提高了17%,ROC AUC从使用PDB 1XL2的0.76提高到使用PDB 1PRO的0.89。这与图4的绿色ROC曲线位于深蓝色ROC曲线左上方、并有大幅的空白间隙是一致的。
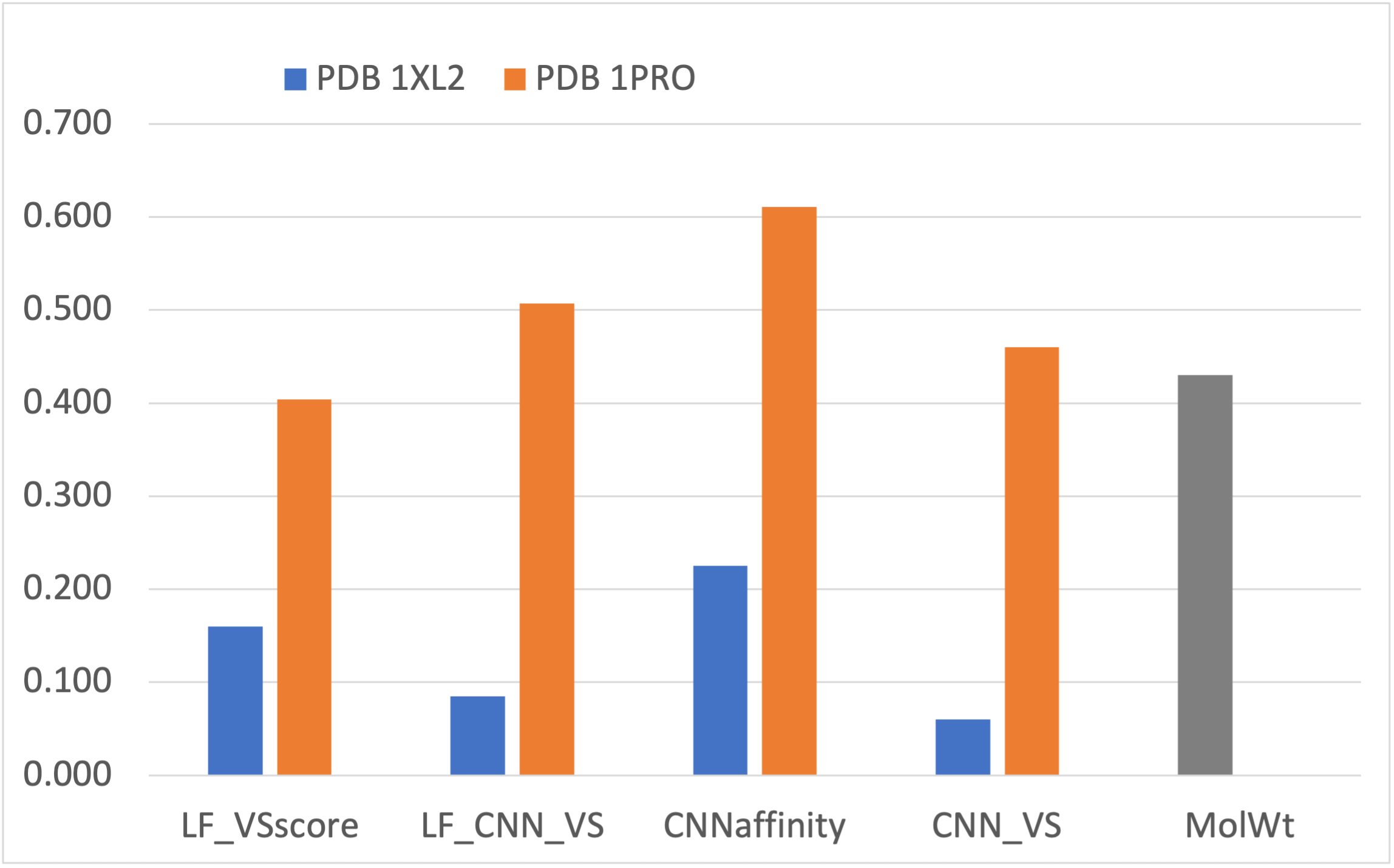
图5. 使用蛋白结构PDB 1XL2与1PRO时的BEDROCα=80.5比较
更重要的是,虚拟筛选的早期富集性能得到大幅度提高。如图5所示,LF_VSscore打分函数的BEDROCα=80.5提高了1.4倍,从使用PDB 1XL2的0.169提高到使用PDB 1PRO的0.404,即使如此,还略低于空模型的0.430;如图6所示,LF_VSscore打分函数的EF1%提高了1.9倍,从使用PDB 1XL2的9.5提高到使用PDB 1PRO的27.6,略高于空模型的26.3。
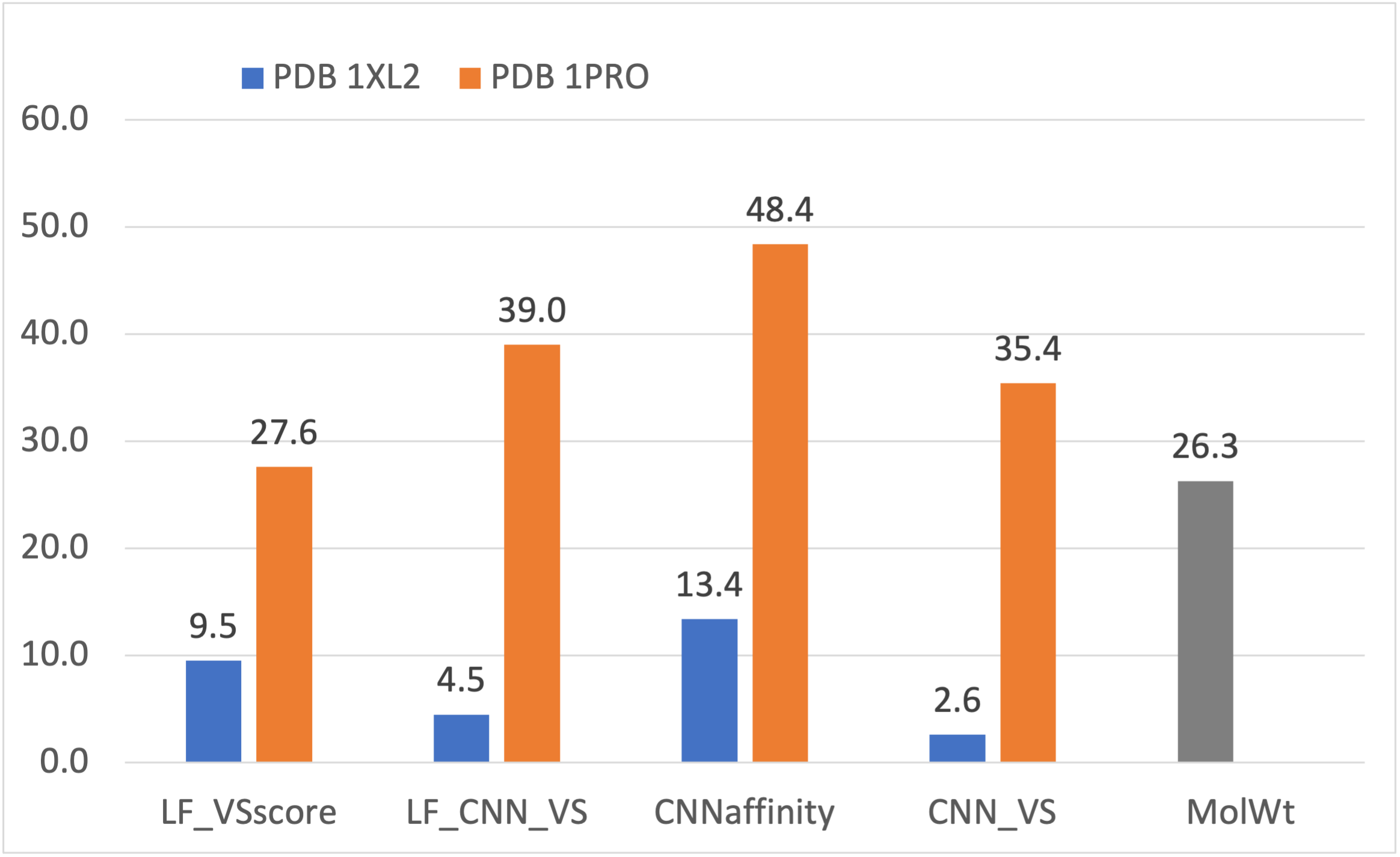
图6. 使用蛋白结构PDB 1XL2与1PRO时的EF1%比较
深度学习CNN affinity打分与数据融合打分CNN_VS、LF_CNN_VS使得虚拟筛选的富集性能显著地优于空模型。具体而言,一方面如图5所示,CNN affinity ,LF_CNN_VS以及CNN_VS三种打分的富集性能指标BEDROCα=80.5分别为0.611、0.507、0.460,优于空模型的0.430;另一方面如图6所示,三种打分函数的富集性能指标EF1%分别为48.4、39.0、35.4,优于空模型的26.3。
根据这个实验,同样的靶标,同样的数据集,我现在会说:Flare docking表现非常优秀,尤其与CNN model数据融合之后更优秀,HIV PR这个靶标也不是很难。
虚拟筛选富集性能与预测的正确pose比例相关:正确比例越高,早期富集性能越好
CNN模型的pose打分(CNNscore)与re-docking以及cross-dock的正确率相关6,打分越高,Pose正确的概率越大。如表5所示,以CNNscore=0.6为截断值,可以发现用PDB 1PRO比用1XL2命中更多的actives化合物数以及更少的decoys化合物数。这与我们推测PDB 1PRO比1XL2的结合口袋与大多数actives分子匹配更好的猜测一致。
表5. 两种蛋白结构虚拟筛选结果包含actives与decoys化合物数
| CNNscore大于0.6 | PDB 1PRO | PDB 1XL2 |
|---|---|---|
| Actives | 281 | 84 |
| Decoys | 2599 | 2948 |
这种正确pose更高比例的对接结果可以转化为更好的虚拟筛选富集能力。用CNNscore对结果进行过滤以模拟这更高pose正确率的子集:将PDB 1PRO虚拟筛选CNNscore小于0.6的对接结果过滤掉,这让我们得到一个理论上高Pose正确率的子集。对过滤后的pose进行性能统计,结果如表6所示。
表6. 使用PDB 1PRO作为蛋白结构并用CNNscore过滤的主要虚拟筛选性能指标
| Metric | LF_VSscore | LF_CNN_VS | CNNaffinity | CNN_VS |
|---|---|---|---|---|
| AUC | 0.741 | 0.741 | 0.742 | 0.740 |
| logAUCλ=0.1% | 53.6 | 51.7 | 53.9 | 49.6 |
| adj-logAUCλ=0.1% | 39.1 | 37.3 | 39.4 | 35.1 |
| BEDROCα=321.9 | 0.679 | 0.678 | 0.757 | 0.575 |
| BEDROCα=80.5 | 0.518 | 0.492 | 0.531 | 0.448 |
| BEDROCα=20.0 | 0.499 | 0.491 | 0.503 | 0.479 |
| EF0.5% | 49.4 | 45.3 | 53.5 | 37.5 |
| EF1% | 37.0 | 32.4 | 37.4 | 27.2 |
| EF5% | 10.0 | 10.1 | 10.0 | 10.0 |
| EF10% | 5.3 | 5.3 | 5.3 | 5.3 |
以LF_VSscore为例,以PDB 1PRO为蛋白结构的虚拟筛选用CNNscore=0.6截断值进行过滤,过滤之后的综合性能有所下降,AUC从之前的0.890(表4)降低到0.741(表6),这是因为过滤使得一部分化合物被排除掉。这一点可以从ROC曲线上可以清楚的看到,如图7所示,经过CNNscore过滤之后的LF_VSscore打分函数ROC曲线(绿色)有一段是直线,这段直线部分是因为过滤造成的,致使绿色曲线比蓝色曲线(未经过滤)的AUC更小。
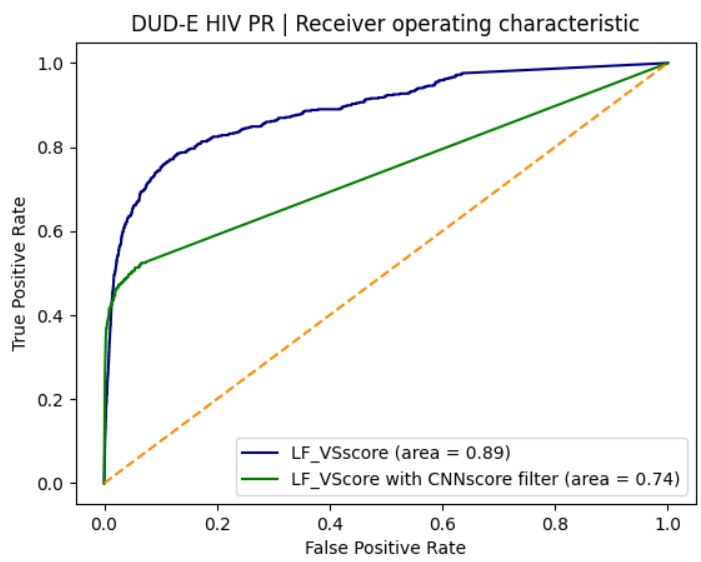
图7. 使用PDB 1PRO作为蛋白结构进行虚拟筛选的ROC曲线。绿色:经过CNNscore=0.6截断值过滤之后LF_VSscore ROC曲线;蓝色:未经CNNscore过滤的LF_VSscore ROC曲线
我们注意到图7绿色曲线比蓝色曲线在非常早期阶段明显地更加靠近纵坐标,这意味着绿色曲线代表的高Pose打分的对接结果具有更好早期富集能力。绘制半对数曲线可以放大ROC曲线早期的细节,结果如图8所示。
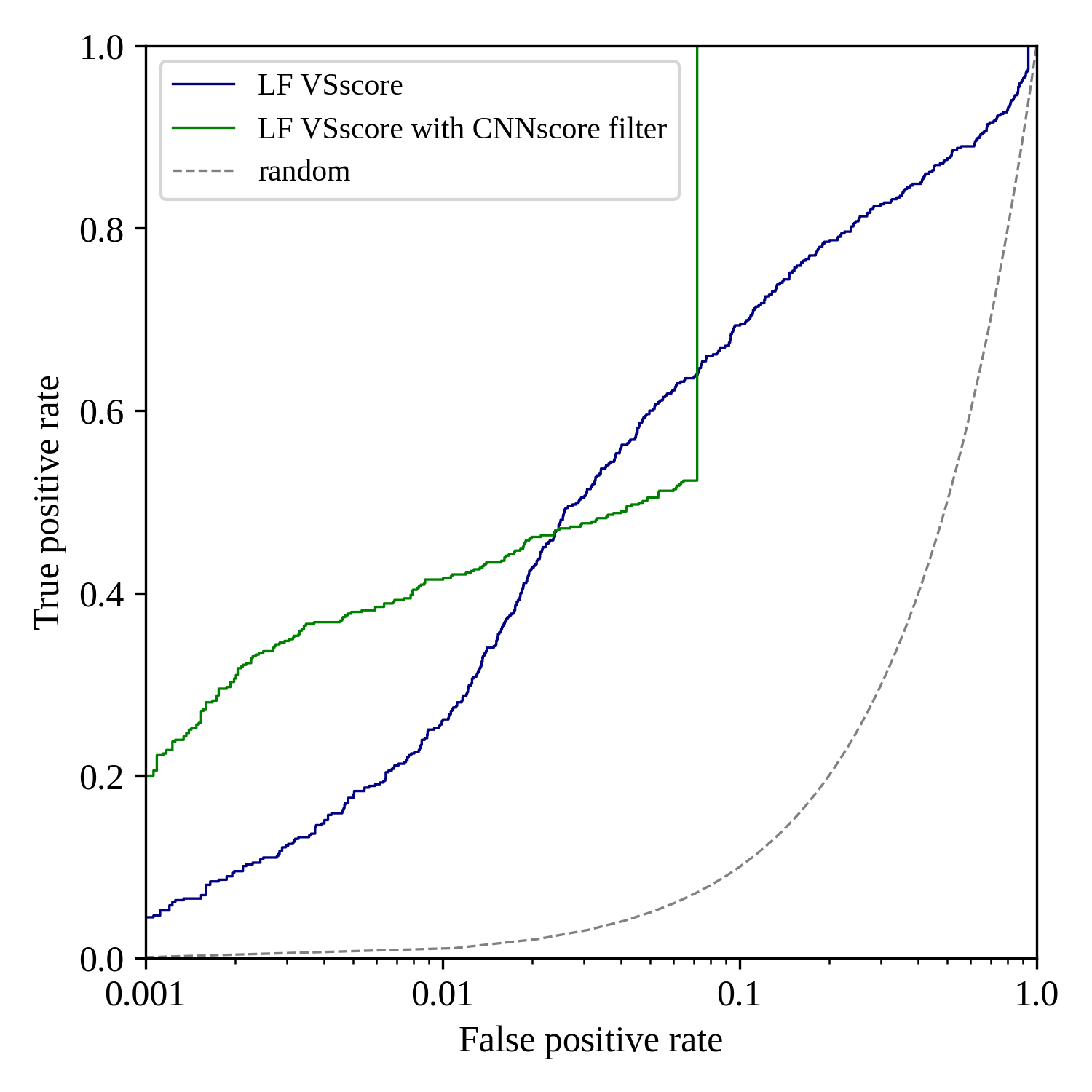
图8. 使用PDB 1PRO作为蛋白结构进行虚拟筛选的半对数ROC曲线。绿色:经过CNNscore=0.6截断值过滤之后LF_VSscore ROC曲线;蓝色:未经CNNscore过滤的LF_VSscore ROC曲线
从图8可以看到,在FPR属于[0.001,0.03]区间的非常早期阶段,绿色曲线(经过CNNscore过滤)位于蓝色曲线(未经CNNscore过滤)的上方,在此阶段,绿色曲线的AUC显著地大于蓝色曲线的AUC,这意味CNNscore过滤得到高概率pose正确率的子集比起全集具有更好的早期富集性能。
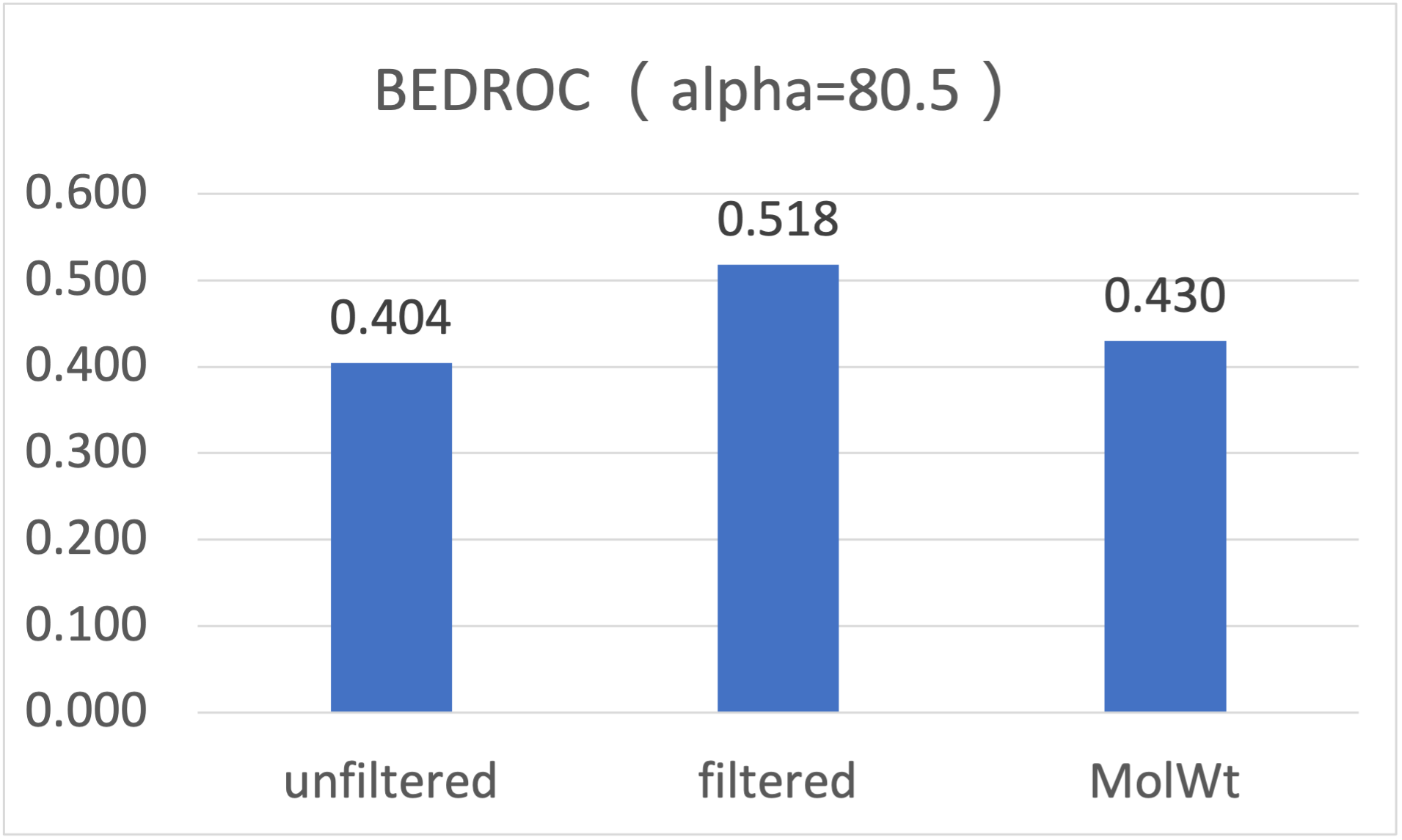
图9. 使用PDB 1PRO作为蛋白结构进行虚拟筛选,经过与未经过CNNscore过滤的早期富集性能指标BEDROCα=80.5比较
除了半对数曲线可以直观看到CNNscore过滤使得LF_VSscore打分函数的早期富集性能得到提高之外,还可以从BEDROC与EF两个定量的富集性能指标看出。一方面,过滤使得早期富集性能指标BEDROCα=80.5提高了28%,如图9所示,从过滤之前的0.404提高到过滤之后到0.518,显著地优于空模型的0.430;另一方面,过滤使得早期富集性能指标EF1%提高了34%,如图10所示,从过滤之前的27.6提高到过滤之后到37.0,显著地优于空模型的26.3。
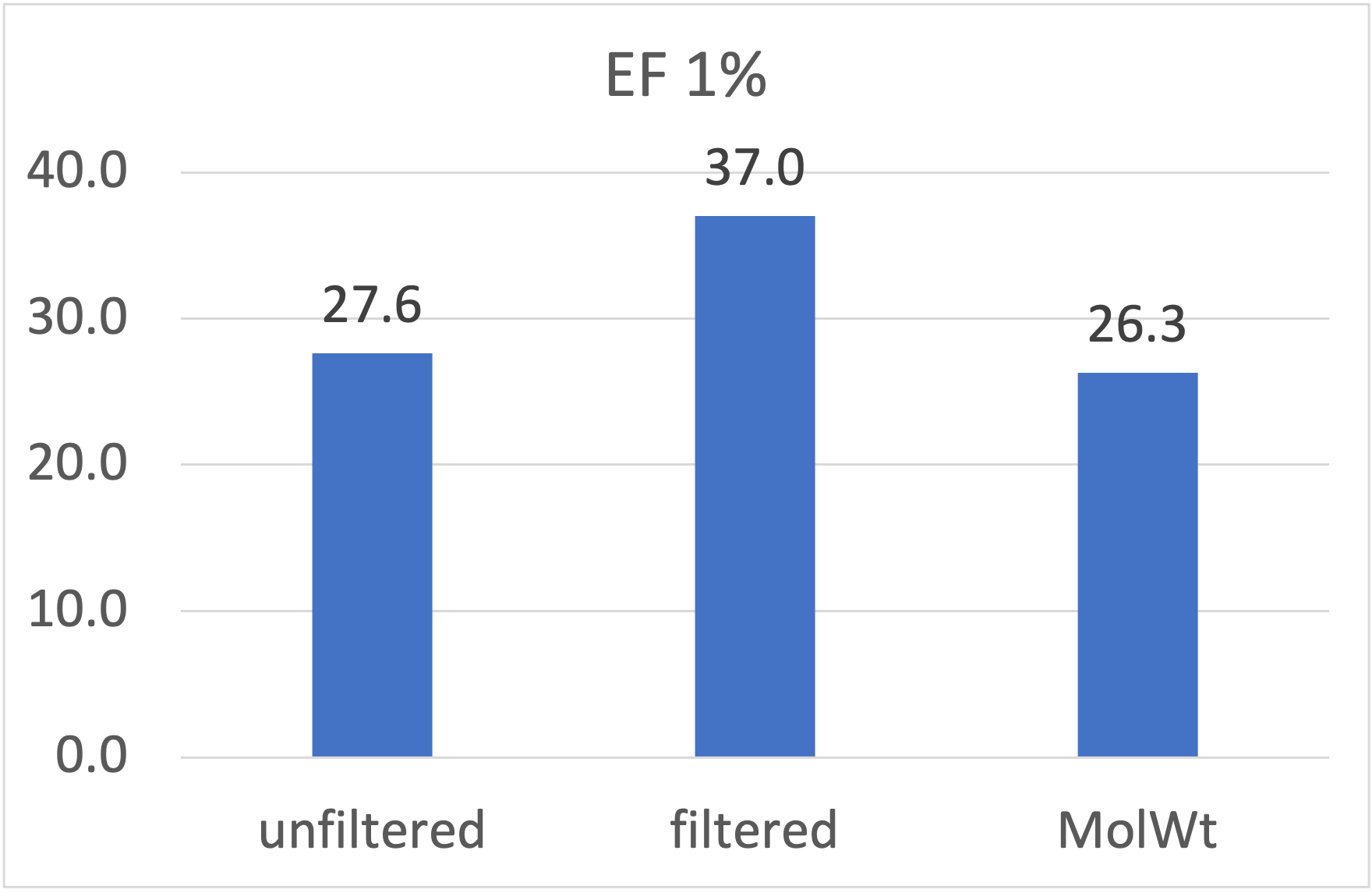
图10. 使用PDB 1PRO作为蛋白结构进行虚拟筛选,经过与未经过CNNscore过滤的早期富集性能指标EF1%比较
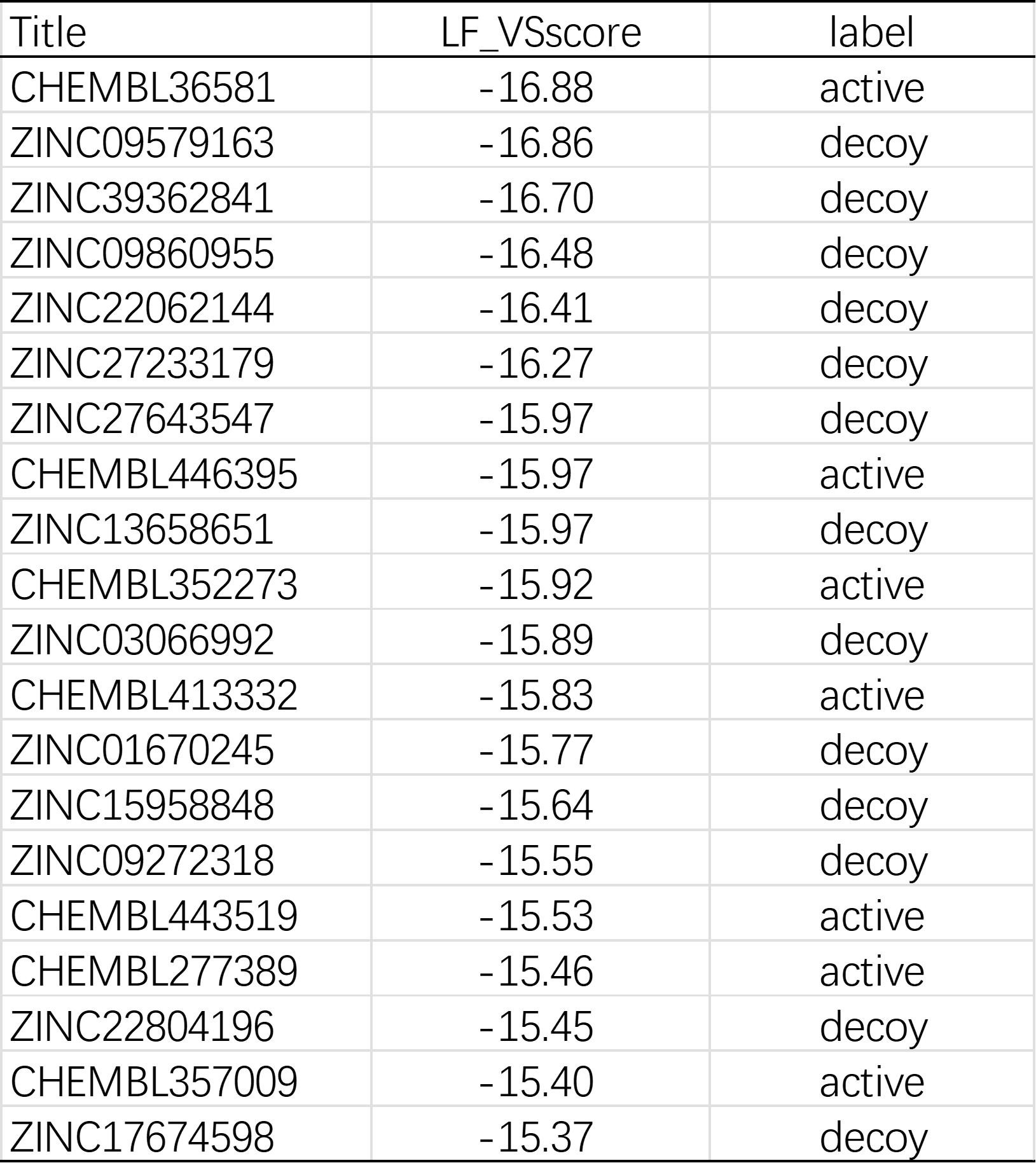
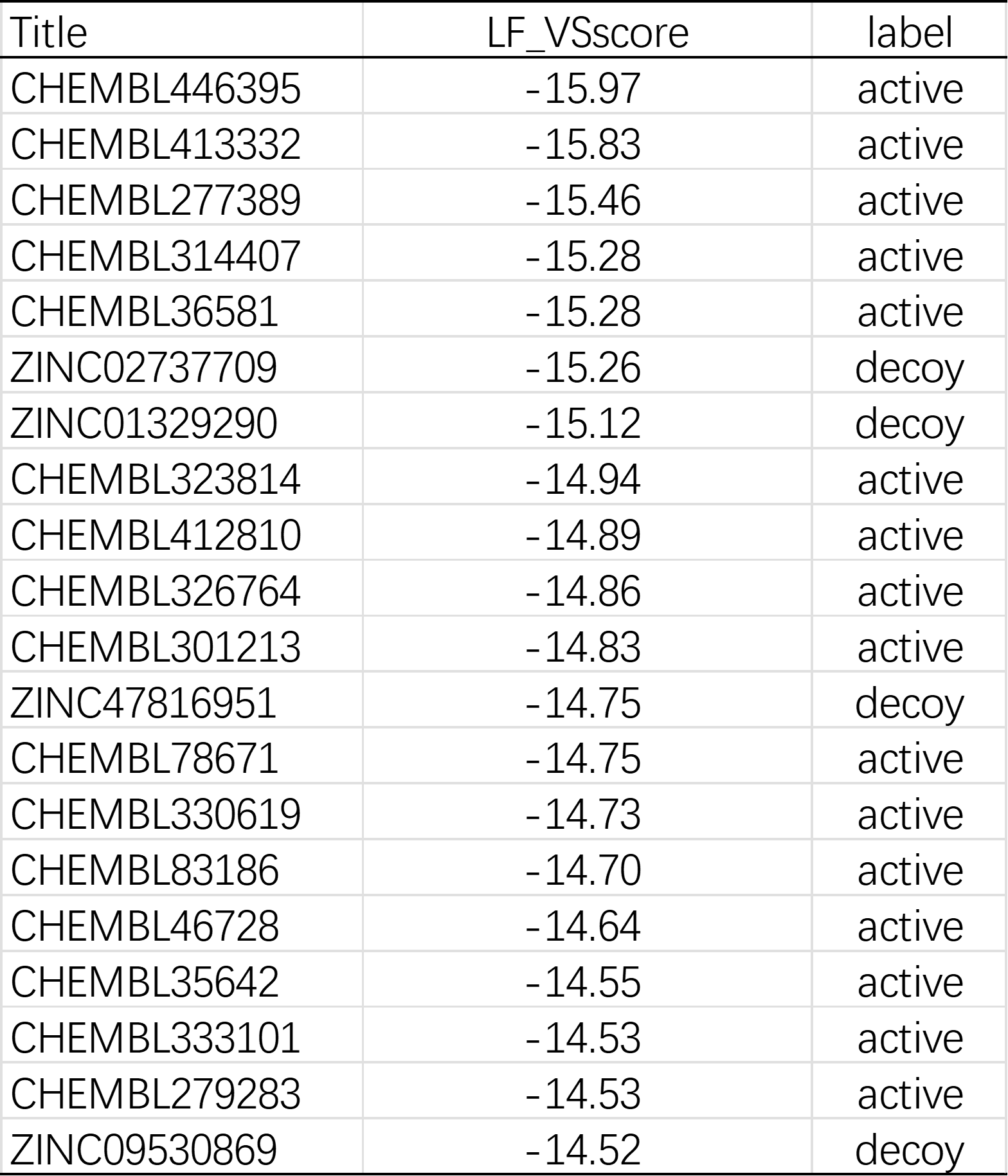
这种理论上更高概率为正确pose的高预测亲和力分子具有更大的机会转化为实验上的高命中率。从实验化学的角度看,在未经过CNNscore过滤的情况下,在LF_VSscore打分排名前20的化合物列表中(表8),已知活性化合物的数量为7;经过CNNscore=0.6的截断值过滤之后,在LF_VSscore打分排名前20的化合物列表中(表9),已知活性化合物的数量提高到16,过滤提高了命中率。
表8. 使用PDB 1PRO作为蛋白结构进行虚拟筛选,LF_VSscore打分最前的20个化合物列表
总的来说,这在个例子中,用CNNscore=0.6的截断值过滤分子对接结果以模拟具有更高正确pose的对接结果子集,结果发现,这种过滤可以提高LF_VSscore打分的虚拟筛选早期富集能力。也就是说,更高正确pose概率的对接结果可以提高打分函数的虚拟筛选富集性能,正确pose的比例越高,早期富集性能越好。选择PDB 1PRO分子对接的蛋白结构比选择PDB 1XL2得到更多高概率正确pose的苗头化合物,因此具有更好的富集性能。
表9. 使用PDB 1PRO作为蛋白结构进行虚拟筛选,经过CNNscore=0.6截断值过滤之后,LF_VSscore打分最前的20个化合物列表
警惕虚拟筛选性能测试的已有结论
我们需要警惕过往论文里关于对不同对接虚拟筛选方法性能排序的结论,这些结论是在非常狭窄的范围内得出,尤其是限定PDB代码的蛋白结构下得出的结论。就如本文的HIV PR这个算例,在用PDB 1XL2蛋白结构时虚拟筛选富集性能很差,在用PDB 1PRO作为蛋白结构之后性能变为非常好。如果你只看到其中性能差的一个,那么得到的性能评测结论是错误的。
以DUD-E的HIV PR子集为例,作者Mysinger等人1,2共分析了110个HIV PR蛋白结构,其中就包含了PDB 1PRO与1XL2,具体的蛋白列表参见原文附件pdb analyze;在蛋白选择(protein selection)步骤实际对多个蛋白结构进行对接虚拟筛选,最后将性能最好的那个呈现给读者,这一点参见原文附件的protein selection,具体内容如下:
Poor overall enrichment. Rebuilt all using biological context. Skipped mutant 1mtb, 1hte has a peptide in the binding pocket, so picked 1xl2 for resolution and enrichment. Tarted GLY27, ASP29, GLY48 and mirrors GLY126, ASP128, and GLY 147. Protonated catalytic dyad ASP124.
作者测试的蛋白结构数量是有限的,有可能没有发现适合用于虚拟筛选的那个蛋白结构。也就是说DUD-E提供的那个蛋白结构有可能并不是最合适的,甚至是差的那个。这提醒我们,在利用文献发表的性能评估结果时,尤其是碰上性能表现差的靶标或方法的时候,在没有尝试更多蛋白结构之前,应该谨慎对待已有的结论。
Wójcikowski等人9将DUD-E数据集分为难的靶标与容易的靶标:如果EF1%小于20则为难的靶标;如果EF1%大于60,则为容易的靶标。结果发现:靶标的难易与数据集中活性化合物的数量没有明显的相关性;难的靶标在很大程度上与所使用的对接软件或机器学习训练方法无关;因此,问题可能是由于受体-配体复合物的3D表示不准确或某些配体的结合位点选择不当。本文的DUD-E HIV PR测试结果为Wójcikowski等人9的推论提供了一个非常好的算例,选择合适的蛋白结构(结合位点)可以显著地改善对接虚拟筛选的富集性能。
结论
在本文中,我们以DUD-E的靶标HIV PR为例,演示了蛋白结构选择对虚拟筛选性能测试的影响。具体的讲,当使用PDB 1XL2作为分子对接的蛋白结构时,Flare docking与文献报道的Glide SP以及GOLD CHEMPLP等的虚拟筛选性能差不多,都没有显著地优于分子量与氢键供体数所代表的空模型的虚拟筛选性能;当使用PDB 1RPO作为分子对接的蛋白结构时,Flare docking表现出优秀的虚拟筛选性能。
本文的实验说明,在同一个靶标上,因蛋白结构选择的不同可导致截然相反的性能评估结论。这警示我们:文献报道的性能评测差的方法不一定真的差,难以虚拟筛选的靶标可能不是真的难,也许只是蛋白结构选择的问题。
在本文中,CNN模型的pose概率打分显示出很强的威力:可以用一致性的数据融合策略,也可以用对接后的pose过滤策略来提高虚拟筛选的早期富集能力。这种能力可能是通过提高正确pose的概率来实现的。选择PDB 1PRO分子对接的蛋白结构比选择PDB 1XL2得到更多高概率正确pose的苗头化合物,因此具有更好的富集性能。
在Flare中,蛋白配体的静电互补性打分(EC score)是另一个评估pose正确性的一个方法。之前的研究8已经表明EC score可以用来指导基于结构的分子设计以及分子对接后处理,在本文中虽然没有讨论,但是相信一定可以与CNNscore进行很好的互补,进一步提高虚拟筛选的性能。
附件
百度网盘下载: https://pan.baidu.com/s/1DpnTU0-nZeOF4xlfQu6Uig
提取码: sr4f
1pro ├── 1pro_ligand.sdf ├── 1pro_prot.grid ├── 1pro_prot.pdb ├── 1pro_prot.pdbqt ├── actives.sdf.gz ├── all_score.csv ├── all_score_grouped.csv ├── decoys.sdf.gz ├── dock.conf ├── metric.csv |
1xl2 ├── 1xl2_ligand.sdf ├── 1xl2_prot.grid ├── 1xl2_prot.pdb ├── 1xl2_prot.pdbqt ├── actives.sdf.gz ├── all_score.csv ├── all_score_grouped.csv ├── decoys.sdf.gz ├── dock.conf ├── metric.csv |
参考文献
- Mysinger, M. M.; Carchia, M.; Irwin, J. J.; Shoichet, B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55 (14), 6582–6594. https://doi.org/10.1021/jm300687e.
- DUD-E HIVPR subset, https://dude.docking.org/targets/hivpr
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of Four Popular Virtual Screening Programs: Construction of the Active/Decoy Dataset Remains a Major Determinant of Measured Performance. J. Cheminform. 2016, 8 (1), 56. https://doi.org/10.1186/s13321-016-0167-x.
- Shen, C.; Hu, Y.; Wang, Z.; Zhang, X.; Pang, J.; Wang, G.; Zhong, H.; Xu, L.; Cao, D.; Hou, T. Beware of the Generic Machine Learning-Based Scoring Functions in Structure-Based Virtual Screening. 2020, 00 (April), 1–22. https://doi.org/10.1093/bib/bbaa070.
- Stroganov et al., Lead Finder: An Approach to Improve Accuracy of Protein-Ligand Docking,
Binding Energy Estimation, and Virtual Screening, J. Chem. Inf. Model. 2008; 48, 2371-2385. - Sunseri, J., & Koes, D. R. (2021). Virtual Screening with Gnina 1.0. Molecules, 26(23), 7369. https://doi.org/10.3390/molecules26237369
- GNINA 1.0, https://github.com/gnina
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein-Ligand Complexes. J. Med. Chem. 2019, acs.jmedchem.8b01925. https://doi.org/10.1021/acs.jmedchem.8b01925.
- Wójcikowski M, Ballester PJ, Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. 2017;7(1):46710. doi:10.1038/srep46710
联系我们获取免费试用
想在自己的项目中用Flare来测试效果,或者获得本文的原始数据,请联系我。


