
摘要:本文分享了如何用深度学习训练分子片段生成器,并提供现成的模型用于分子片段生成,还分享了分子片段生成器在药物设计领域的应用场景。并以少量含能材料分子进行迁移学习训练,发现新的模型确实可以生成我们想要的结构类型化合物;此外,还分享了深度学习用于生成EGFR激酶抑制剂类药分子的生成与光伏材料分子的生成。
作者:肖高铿
日期:2019-02-24
背景: 从头设计
AutoT&T与Ligandscout药效团虚拟筛选联用
在我的博客文章《Ligandscout教程 | 基于片段的虚拟筛选联用AutoT&T进行从头设计》中已经展示过AutoT&T与Ligandscout一起使用进行分子片段替换自动生成分子结构的设计策略。Ligandscout可以快速地对片段库进行虚拟筛选,然后AutoT&T从命中的片段库中进一步按键匹配性将片段裁剪、拼接到先导化合物上,实现侧链替换或骨架跃迁。在该例子中,Ligandscout的片段虚拟筛选与AutoT&T结合快速地生成打分优秀的全新CDK2分子。
AutoT&T与分子对接虚虚拟筛选联用
AutoT&T还可以与任意的分子对接软件,比如Flare,联合使用实现多起点优化策略:将已知抑制剂对接到结合位点里,再用AutoT&T将pose进行裁剪、拼合实现全新分子设计。这个策略的好处是:可以利用别人成功发现的高活性分子进行片段拆解,重新组合出新的化合物,这些化合物可能具有很高概率具有良好的活性。在AutoT&T 2.0算例 | VEGFR-2抑制剂的多起点优化博客文章中,展示了如何联合使用AutoT&T与GOLD实现这个“多起点优化策略”生成全新潜在的VEGFR-2抑制剂。当然,分子对接可以对片段进行虚拟筛选,再用AutoT&T进行高效的从头设计。为了便于大家使用这个策略,我们特意为学术用户推出了Flare学者版及其与AutoT&T捆绑特价套餐,欢迎咨询我们。
SPARK: 基于配体的从头设计
SPARK是一款用户非常友好的全新分子设计软件,最新版本为10.5,除了骨架跃迁与侧链替换之外,还包括:1)分子生长;2)片段链接;3)分子内环合;4)结合位点水分子替换;5)基于药效团的约束;6)将新化合物发送给Flare进行基于结构的计算评估等等。SPARK包含一整套预先准备的片段库,其中部分片段库每周更新,为你带来最大化的药物发现机会。
深度学习从头设计(自动生成分子结构)
深度学习在近几年非常热门,已经有非常多的综述。而我最关心其中两个应用:
- 建立预测性QSAR模型:回归与分类
- 自动结构生成:从头设计
预测化合物的理化性质、毒性、活性等等。比如Merck公司的Yuting Xu、Junshui Ma等人的DeepNeuralNet QSAR项目1,2就涉及回归模型。DeepChem专门设计用深度学习解决化学问题,有非常多回归与分类方面的应用例子。
即机器自动生成新的结构或类似物。比如阿斯利康的Olivecrona等人的REINVENT3,4项目以及Mariya Popova等人的ReLease12,13项目涉及深度学习进行化合物从头设计,以及在低数据量下的迁移学习生成感兴趣的化合物。Max Hodak7,8, Esben Jannik Bjerrum10与Novartis的Peter Erlt等人11分享了用深度学习实现从头设计的经验。

Figure 1. REINVENT迁移学习自动生成Celecoxib衍生物(图片来源自REINVENT项目网站4)
除了生成类药分子,还可以生成类生源化合物。比如中山大学药学院徐俊教授课题组的Shuangjia Zheng与Xin Yan等人在REINVENT的基础上,进一步开发了QBMG4,5:(1)用ZINC生源性(biogenic)化合物训练深度学习模型,QBMG可以成功的生成类似于天然产物样分子;(2)通过迁移学习,对特定类型的化合物进行训练,可以生成特定类型的天然产物类似物。
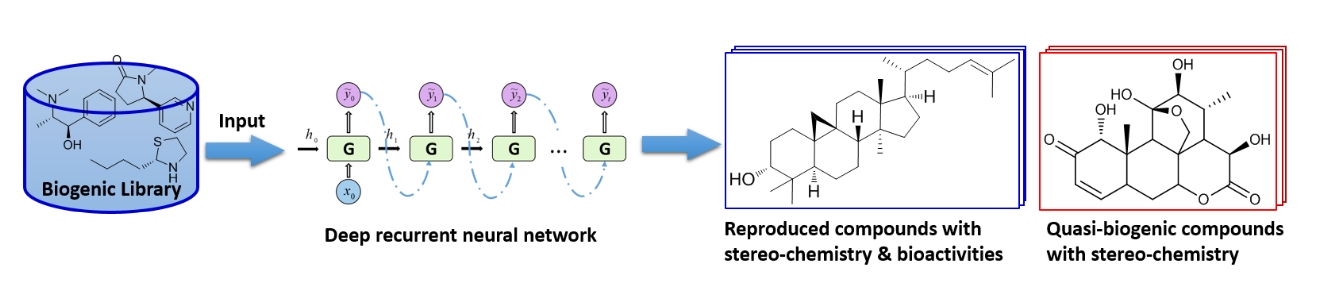
Figure 2. QBMG-深度学习准生源化合物生成器(图片来源自QBMNG项目网站6)
深度学习结构生成器与传统药物设计方法的结合
我最感兴趣的还是如何将深度学习用于药物设计。REINVENT提供了一个思路:将结构生成器与QSAR/打分函数联用,结构生成后用各种预测工具进一步对化合物进行过滤,最后获得性质最优的全新化合物。只要是支持命令行的工具比如3D-QSAR(CoMFA,Forge的Field-based 3D-QSAR),各种虚拟筛选工具(比如分子对接虚拟筛选,药效团虚拟筛选),性质计算工具(比如xlogp)都可以在源代码层面或命令行使用的方式与深度学习结构生成器直接联用、输出目标化合物。但这并不是必需的方式,可以将结构生成器获得的结构保存,手动分步骤方式一步一步用QSAR、虚拟筛选、性质计算等方式进行进一步评估、观察与决策。
除了将深度学习用于类药分子生成之外,我还对生成分子片段感兴趣,理由:1)基于片段的分子设计具有独特的优势,已经成熟地应用于药物发现与优化;2)片段分子具有更小的原子数组成,更适合于深度学习、训练结构生成模型;3)最重要的是,我有很多优秀的基于片段的药物设计软件(SPARK,Ligandscout,Flare与AutoT&T),可以快速地获得分子设计灵感,这些灵感具有人类所构思不出来的结构片段。
基于深度学习的片段生成(Deep learning based fragment generator, DLFG)
我fork了一个QBMG9,并将训练好的片段模型复制到QBMG的data目录。现在,你可以直接用QBMG直接生成片段分子了。我生成了20000个分子,虽然有“键”不合理的化合物,但大部分看起来确实是片段样化合物,下图是前25个分子的2D结构。
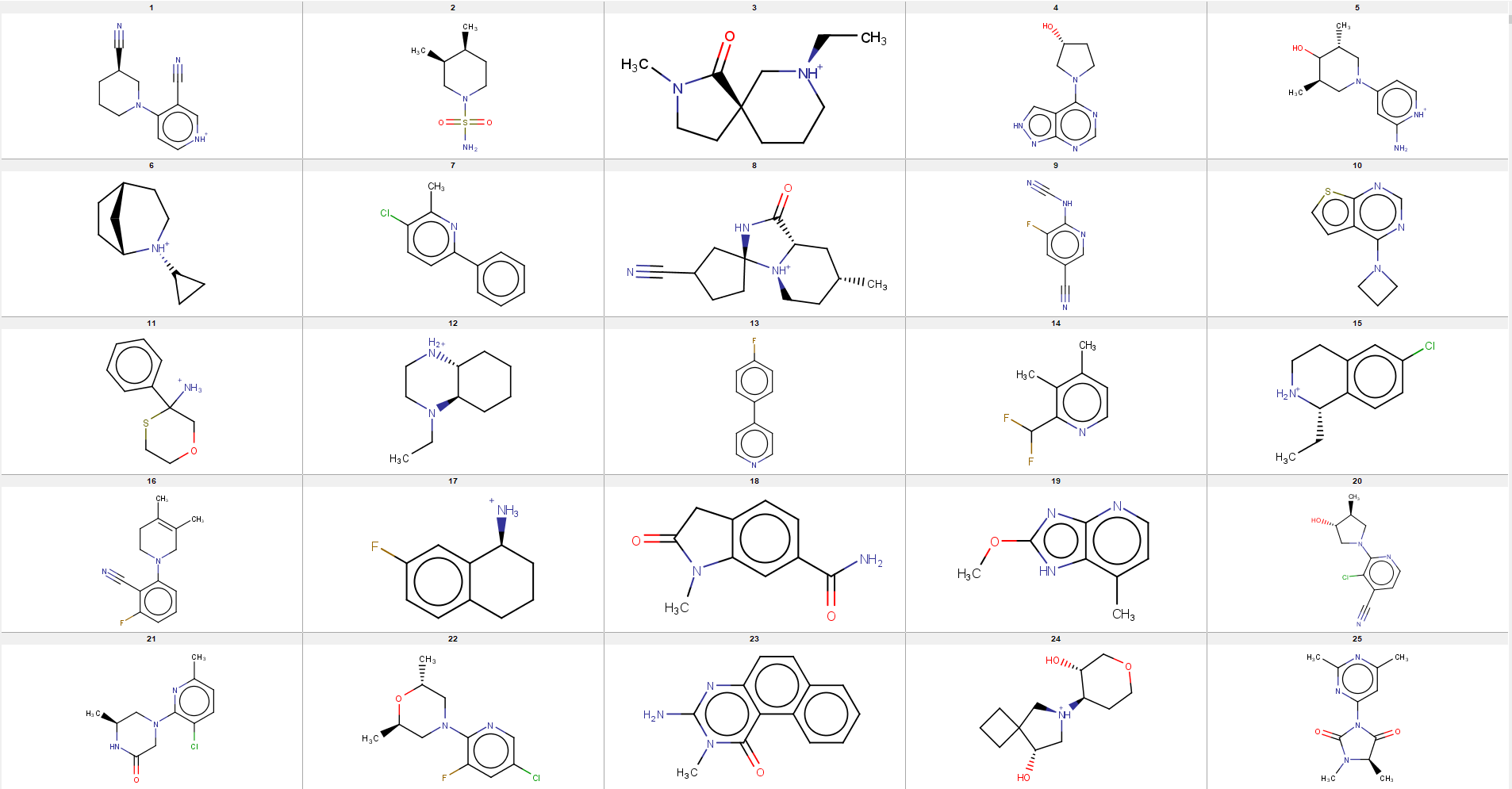
Figure 3. 部分片段生成器生成的化合物
DLFG结构生成器源代码与实例
含能材料分子生成
硝基在ZINC数据库里并不常见,而常见于含能材料分子(比如炸药)。通过对富含硝基的化合物进行迁移学习可以很容易地观察训练集化合物对结果的影响。手头并没有含能材料分子,于是从南京理工大学钱博文(2016)硕士论文《含能材料的撞击感度等等安全参数的定量构效关系研究》里画了20多个硝基化合物。还从ZINC里提取100多个氧平衡值大于-50的化合物作为训练集化合物,虽然这些化合物不是含能化合物,暂且就当成是:主要是观察经过迁移学习后新的模型能否生成多硝基类化合物。结果表明,模型确实可以生成多硝基化合物,部分化合物的氧平衡值高达40多。虽然这只是一个简单的测试,生成的结构也不理想,需要更多的化合物进行训练。我们可以明显地看到化合物的氮含量与硝基增加,与之前的化合物有明显区别,这表明迁移学习确实可以训练获得我们想要的化学结构类型。
激酶抑制剂结构生成
进一步尝试用迁移训练药物分子生成,分别用1000多个EGFR抑制剂进行迁移学习训练,结果如下。
- EGFR激酶
点击下载生成的结构:egfr_denovo.csv
Table 1. 部分生成的EGFR激酶结构
|
|
|
|
|
|
浏览这些化合物,会发现有少部分结构显然与靶标不相干,这说明需要进一步用分子对接、QSAR、药效团等对化合物进行进一步的过滤、筛选。同时,也有部分化合物结构不合理,比如EGFR激酶结构生成的EGFR_TF_356(Figure 4),需要进一步过滤去除。但是,错误的结构也并非没有用处,你依旧可能从中得到提示:如果这个化合物有可取之处,你简单地修改就可能得到好的创意。
Figure 4. 在EGFR激酶生成中一个不合理键的例子:EGFR_TF_356结构中有个不合理的N-O键。
光伏材料分子生成
同样道理,我们还可以对现有的光伏材料分子进行训练生成类似光伏材料的分子,Table 2随机给出部分生成的结构。
点击下载生成的部分结构:opv.smi
Table 2. 深度学习生成的部分OPV分子结构
|
|
|
|
|
|
对深度学习与传统技术的一点看法
传统的从头设计(比如LigBuilder,EA-Inventor)开发不仅要掌握化学、还需要懂编程,需要经过几年、十几年的艰苦训练才能开发出一套实用的从头设计软件。深度学习是大数据时代的产物,只要有大量的数据就可以让不懂药物的IT开发者、普通的药物化学工作者经过简单的培训就可以快速地开发出结构生成模型,并获得感兴趣的化合物结构。比如,只要提供足够数量的EGFR激酶,简单训练就可以让软件自己设计化合物。
从掌握结构生成技术难易程度角度讲,深度学习比传统技术具有无与伦比的优势。但这并不意味着深度学习就可以颠覆药物开发这个行业。深度学习确实让人摆脱了为掌握编程而所需的长时间、艰苦的学习训练过程,但是深度学习依赖于传统方法产生的数据,同时也需要结合传统技术才能得到更可靠的结果。与深度学习相比,掌握传统从头设计开发方法并不容易,到目前为止也只有几个课题组与公司掌握这个技术。即使有从头设计开源软件,不会编程也难以按自己的想法修改。但是,传统技术的从头设计方法却不依赖于数据就可以生成有创意的结构,比如直接从蛋白结合位点从无到有的长出化合物结构。我认为掌握方法的难易程度与对数据的依赖程度是深度学习与传统技术的最大区别之处。
直觉认为材料领域将会是深度学习数据依赖的例外领域,比如光伏材料(太阳能电池)与OLED领域。因为这些领域早期化合物筛选的关键指标可以通过理论计算获得,也就是说只要计算能力足够,就可以源源不断地通过计算制造数据并用于深度学习训练、设计新的化合物,再对新的化合物进行性质计算,再返回去训练模型。基于此,能源、OLED领域将更容易从深度学习中获益。
接下来可以做什么
- 用迁移学习训练特定结构类型的片段生成器:比如激酶骨架生成器。
- 与Ligandscout与LeadFinder,AutoT&T联用,设计全新的分子: 对现有片段库进行补充,希望从深度学习中获得意想不到的新骨架。
- 直接作为SPARK的片段数据库进行基于配体的骨架跃迁与片段替换。
- 期待你的意见,寻求合作机会。
文献
- Xu, Y.; Ma, J.; Liaw, A.; Sheridan, R. P.; Svetnik, V. Demystifying Multitask Deep Neural Networks for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2017, 57 (10), 2490–2504. https://doi.org/10.1021/acs.jcim.7b00087.
- Yuting Xu; Junshui Ma. DeepNeuralNet-QSAR. https://github.com/Merck/DeepNeuralNet-QSAR, accessed 2019-02-24
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular De-Novo Design through Deep Reinforcement Learning. J. Cheminform. 2017, 9 (1), 48. https://doi.org/10.1186/s13321-017-0235-x.
- Olivecrona, M. REINVENT. https://github.com/MarcusOlivecrona/REINVENT. accessed 2019-02-24
- Zheng, S.; Yan, X.; Gu, Q.; Yang, Y.; Du, Y.; Lu, Y.; Xu, J. QBMG: Quasi-Biogenic Molecule Generator with Deep Recurrent Neural Network. J. Cheminform. 2019, 11 (1), 5. https://doi.org/10.1186/s13321-019-0328-9.
- SYSU-RCDD. QBMG: Quasi-Biogenic Molecule Generator with Deep Recurrent Network. https://github.com/SYSU-RCDD/QBMG. Accessed 2019-02-24
- Gómez-Bombarelli, R.; Wei, J. N.; Duvenaud, D.; Hernández-Lobato, J. M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T. D.; Adams, R. P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4 (2), 268–276. https://doi.org/10.1021/acscentsci.7b00572.
- Max Hodak. Keras molecules. https://github.com/maxhodak/keras-molecules.accessed 2019-02-24
- Gaokeng Xiao. Use QBMG to generate fragment-like molecules. https://github.com/gkxiao/QBMG. Accessed 2019-02-24
- Esben Jannik Bjerrum.(2017-12-14) Master your molecule generator:seq2seq rnn models with smiles in keras. https://www.wildcardconsulting.dk/master-your-molecule-generator-seq2seq-rnn-models-with-smiles-in-keras. Accessed 2019-02-24.
- Ertl, P.; Lewis, R.; Martin, E.; Polyakov, V. In Silico Generation of Novel, Drug-like Chemical Matter Using the LSTM Neural Network. 2017, arXiv:1712.07449
- Popova, M.; Isayev, O.; Tropsha, A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018, 4 (7), eaap7885. https://doi.org/10.1126/sciadv.aap7885.
- Olexandr Isayev.ReLeaSE (Reinforcement Learning for Structural Evolution).https://github.com/isayev/ReLeaSE. Accessed 2019-02-24


