
用SPARK重现选择性大环CDK2抑制剂QR-6401的设计
摘要:本文以一个“线型”的CDK2抑制剂化合物13为起点分子,经过分子对接预测结合模式,并用SPARK大环化工作流成功地设计出文献报道的大环CDK2抑制剂QR-6401以及其它化学结构类型的五原子长度连接臂的大环化合物。除此之外,还包含许多打分高但没有被文献报道的新类型化合物。Spark大环化比深度学习生成大环分子更加高效,比如在Spark大环化计算实验中直接设计出QR-6401,在深度学习生成的结构中并不包含QR-6401,而是在先导化合物基础上由药化专家修饰得到。
肖高铿/2023年2月23日
前言
选择性CDK2抑制剂有可能为CDK2依赖性癌症提供有效的治疗方法,并对抗由于细胞周期蛋白E1(CCNE1)高表达或CDK4/6抑制剂治疗诱发的CCNE1扩增而产生的耐药性。腾讯与齐鲁锐格的Yu等人1最近报道了通过生成模型和基于结构的药物设计(SBDD)加速发现了一种高效、选择性的大环CDK2抑制剂QR-6401(化合物23,见图1),QR-6401在OVCAR3卵巢癌异种移植模型中表现出强大的口服抗肿瘤效果。
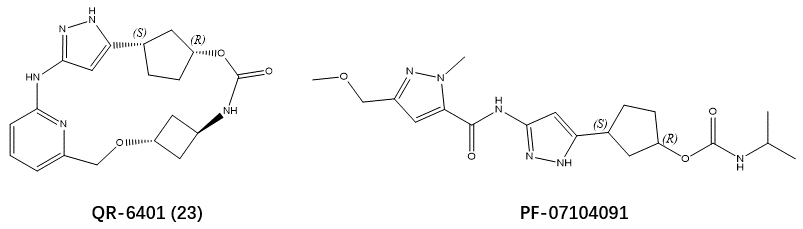
图1. QR-6401(化合物23)与PF-07104091的化学结构式
QR-6401是以“线型”分子13为起点用MacroTransformer生成而来,大环化的策略如图2所示:一个连接点在13的吡啶环6位碳上,另一个连接点在另一端的酰胺氮上,连接臂(linker)长度设置为4-6个原子。大环化策略得以使用是基于在13与CDK2的结合构象中两个连接点原子紧挨着这个事实:它们之间的距离为5.2Å。
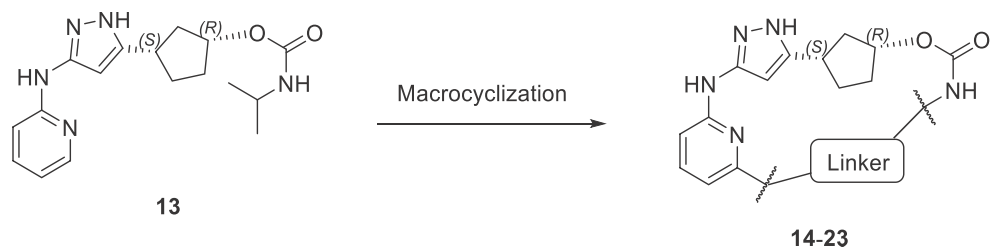
图2. Yu等人1的大环化化合物生成策略
Yu等人1的MacroTransformer计算实验生成了7626个大环化合物。这些化合物进一步用基于Xed力场的场点技术2,3以13为参比分子进行形状与场相似性打分,保留场点相似性大于0.65的化合物978个。保留下来的化合物进一步用glide对接到化合物13的结合位点里,这一步的对接虚拟筛选获得了792个化合物共30种化学类型。最后药化科研从中精心选择了10个化合物进行了合成与活性测试,结果如表1所示。
表1. MacroTransformer生成、人工精选后的大环化化合物及其活性
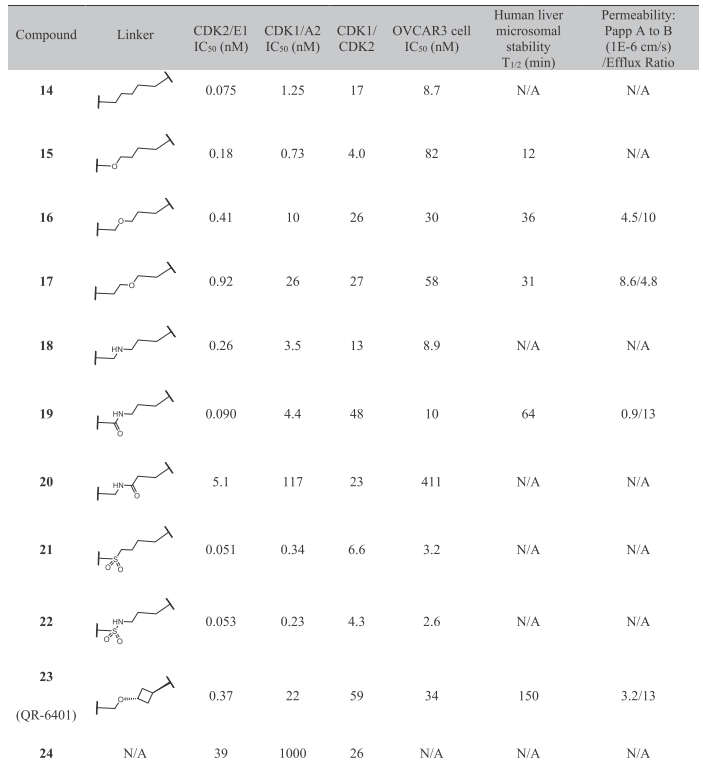
从表1可知,这些精选化合物的共同特点是连接臂长度为5个原子,除了化合物20之外,其它的化合物都比线型化合物13(CDK2/E1 IC50=8.1 nM)的酶学活性提高了至少1个数量级,其中QR-6401(化合物23)的人肝微粒体代谢稳定性也大幅度提高,实现了大环化技术在提高活性、改善DMPK性质方面的优势。为了验证这些活性的提升确实为大环化所带来的,作者还设计了没有连接臂的化合物24,不出所料,24的活性比大环化合物活性严重降低。
本文的目的是以化合物13为起点分子,尝试用SPARK4大环化工作流重现Yu等人1从13到QR-6401的发现过程,并了解Spark大环化工作流产生的其它令人感兴趣的化合物。
方法
起点分子13的结构准备
起点分子正确的生物活性构象是Spark计算实验的关键,通常来源于配体-蛋白复合物结构。虽然Yu等人1已经解释了CDK2与化合物13(PDB 8H6T)与19(PDB 8H6P)的共晶结构,但是在撰写本文的时候还处于未公开状态。化合物13衍生自PF-07104091,该化合物也没有共晶结构可供使用(图1)。鉴于此,有必要预测13的生物活性构象,
截止目前(2023年2月23日),在PDB上公开了429个X-衍射方法解释的CDK2晶体结构,这些结构均包含ATP结合的结构域。下载这些结构,并用Flare进行蛋白结构准备,序列比对,以PDB 1GIJ为模板,并按Cα拟合进行3D叠合,这么做的原因是确保仅用1GIJ的共晶配体也可以定义apo结构的结合位点。接下来将化合物13用Flare Docking对接到每个结合位点(共670个结合位点),对接时仅将蛋白链作为受体,水、金属、以及其它的成分均被忽略。结果表明,化合物13打分最优的是PDB 4FKU的配体结合位点,并与Yu等人1描述的相互作用模式比较一致。因此,进一步以PDB 4FKU的蛋白链作为受体,在Very accurate but slow模式下进行对接获得结合模式。
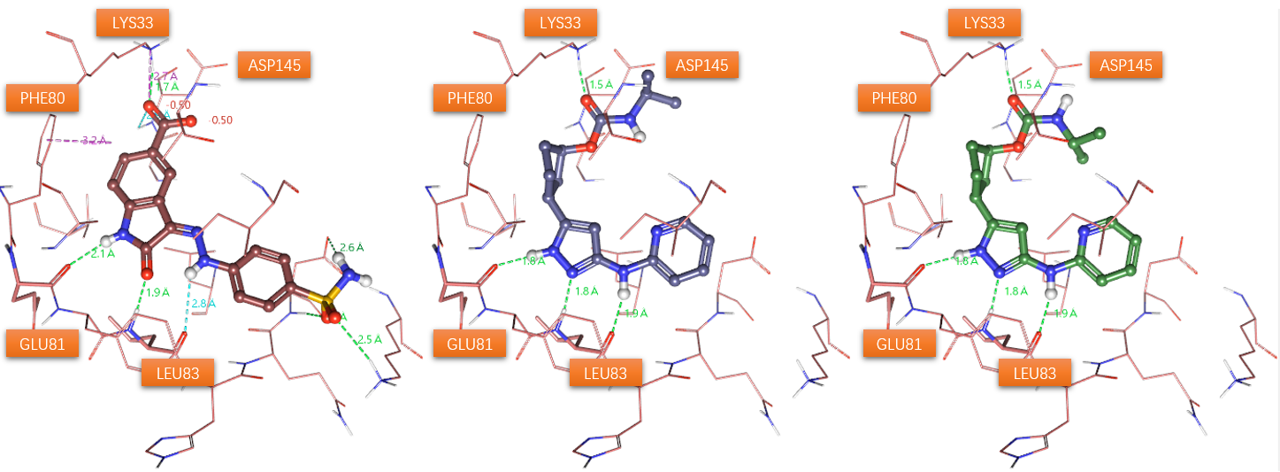
图3. 分子对接预测的化合物13在PDB 4FKU配体结合位点里的结合模式。左:PDB 4FKU共晶配体;中:预测的化合物13的结合模式;右:手动将化合物13酰胺键旋转为顺式后的结合模式
分子对接预测化合物13的结合模式如图3(中)3D图与图4相互作用2D图所示:吡啶氨基作为氢键供体与LEU83的羰基发生氢键相互作用,吡唑氮受体与LEU83的酰胺NH发生氢键相互作用,吡唑NH作为供体与GLU81的羰基发生氢键相互作用,环戊烷主要与PHE80发生很多的疏水接触,碳酰胺羰基氧作为受体与LYS33末端的阳离子发生氢键相互作用。这些相互作用模式与Yu等人1描述的基本一致。
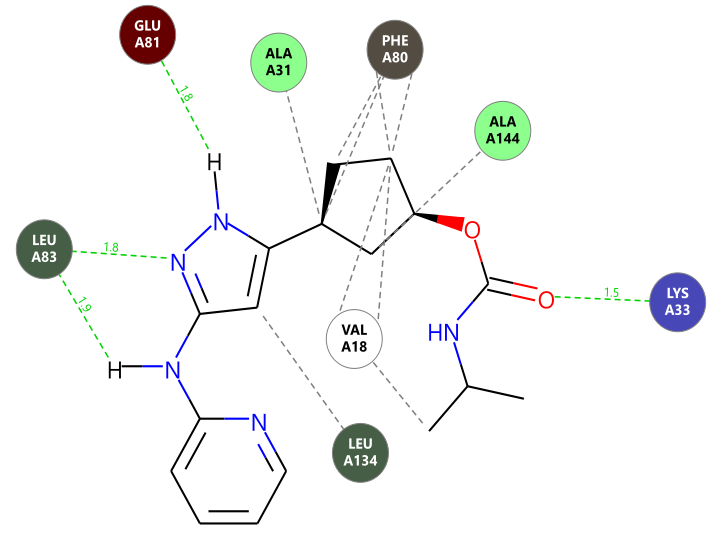
图4. 分子对接预测的化合物13的结合模式2D图
预测的结合模式与Yu等人1报道共晶结构(PDB 8H6T)存在一个显著差异是碳酰胺片段的构象:在本次对接预测中碳酸酰胺的羰基氧与NH为反式,而在共晶结构中则为顺式。一般来说顺式酰胺比反式酰胺具有更高的构象能,用Flare的QM模块在B3LYP-D3BJ/6-311G(d,p)理论水平对13的碳酸酰胺片段O=C-N-C进行两面角扫描,结果如图5所示,顺式构象(180°)比反式(0°)构象的能量高了1.2kcal/mol。在共晶结构PDB 8H6T中,13的顺式碳酸酰胺NH作为氢键供体还与ASP145的末端羧酸发生氢键相互作用,这对高能的顺式构象具有稳定化的作用。而在对接的蛋白结构PDB 4FKU中,ASP145距离碳酸酰胺片段的NH较远,不能与酰胺NH发生氢键相互作用,在这种情况下内能更低的反式构象是更有利的结合模式。这也进一步说明了有必要对化合物13进行大环化的构象约束以锁定相互作用有利的高能构象以提高结合亲和力。
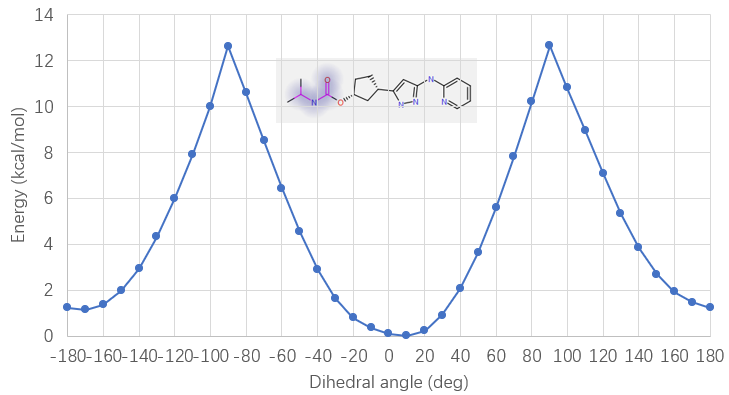
图5. 化合物13碳酸酰胺片段在B3LYP-D3BJ/6-311G(d,p)理论水平下两面角扫描
为了与PDB 8H6T共晶配体构象一致,在结合位点里将对接结果的酰胺键手动旋转为顺式,得到如图3-右所示的结合模式作为下一步大环化的起点分子。这里,我将两个典型的可能结合模式以Flare项目文件以及通用文件的形式提供下载:13-binding-mode.flr,13-binding-mode.zip。它们之间的区别在于:其中一个结合模式的碳酸酰胺片段、吡唑片段、氨基吡啶片段的原子几乎处于同一个平面,此时碳酸酰胺片段的N原子与吡啶6位C原子之间的距离为5.7Å;而另一结合模式的吡啶环稍偏离平面,围绕氨基-吡啶的C-N旋转了大约8°,此时碳酸酰胺片段的N原子与吡啶6位C原子之间的距离为5.2Å。我认为这两个构象均可作为活性构象用于下一步的大环化设计,具体的讲,我选择了用更加平面化的那个构象作为Spark大环化设计的起点分子。
化合物13的大环化设计
SPARK从片段数据库搜索具有相似静电和立体特性的片段来替换给定起点分子(starter)中的特定部分6, 其中静电和立体特性的相似性比较是基于XED力场计算得到的分子场来计算2,7-8。SPARK在搜索可替换片段时不仅对相应的替换片段进行评分,而且还对分子整体进行评分。替换片段是从SPARK的片段数据库中筛选出来的,是从可供商品销售的化合物数据库或文献报道的化合物生成而来。当一个片段被识别为含有所需数量的连接点并具有与起点分子相似的形状/几何形状的片段之后,SPARK会用XED力场对分子进行能量最小化计算以去除不良的碰撞和不利的几何形状,然后再进行最终分子的场计算和相似性优化以得出最终的打分值。
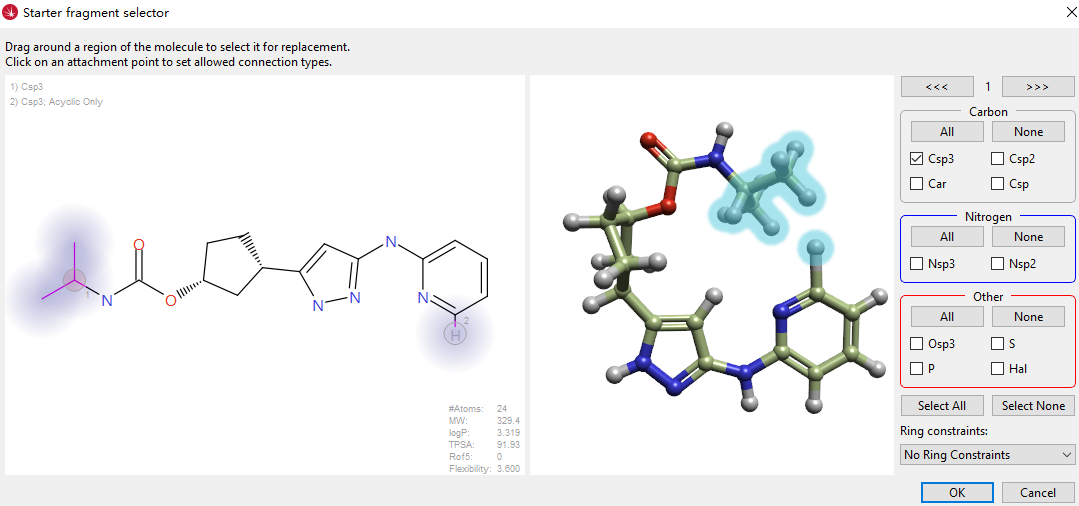
图6. 大环分子设计的起点分子以及连接点。左边高亮部分是连接点:连接点1是碳酸酰胺氮上的异丙基;连接点2是吡啶6位碳上的氢原子。右边的3D视窗可以实时观察连接点以确保连接点的选择是正确的。
SPARK通过的分子大环化向导来控制大环化合物的生成策略。大环化向导模块的图形界面可用来进行分子大环化设计的参数设置(图6)。 在本实验中,起点分子即为上一步预测的化合物13的结合构象,大环化策略如图6所示:将碳酸酰胺氮上的异丙基设置为连接点1,将吡啶环6位碳上的氢原子设置为连接点2,连接点1与连接点2的原子均设为sp3杂化的碳原子,且没有其它的化学类型要求。

图7. 大环化设计使用的片段数据库
本次实验使用“Ligand Joining / Macrocyclization”设置参数来选择需要筛选的片段数据库。如图7所示,本次实验对文献数据库ChEMBL以及Commercial数据库的全部子库进行了筛选,具体而言,包括了下列数据库:
- ChEMBL_common
- ChEMBL_rare
- ChEMBL_veryrare
- Commercial_VeryCommon
- Commercial_Common
- Commercial_LessCommon
- Commercial_Rare
- Commercial_VeryRare
- Commercial_ExtremelyRare
- Commercial_UltraRare
虽然Yu等人1在大环化过程中对连接臂定义了长度为4-6个原子,然而在本研究中我们不设任何限制,所有的其它选项均采用默认值。
结果
本次计算实验共生成500个大环分子,对连接臂(linker)用Bemis-Murcko clustering方法进行聚类分析,总共包含146种不同化学类型,比Yu等人1用MacroTransformer生成30种化学类型要多。
Spark除了生成更多的化学结构类型之外,其打分分布也更高。如图8所示,这些大环分子与起点分子13相比具有非常高的形状相似性与场相似性,其中形状相似性在0.875-0.950之间,而场相似性在0.817-0.923之间。
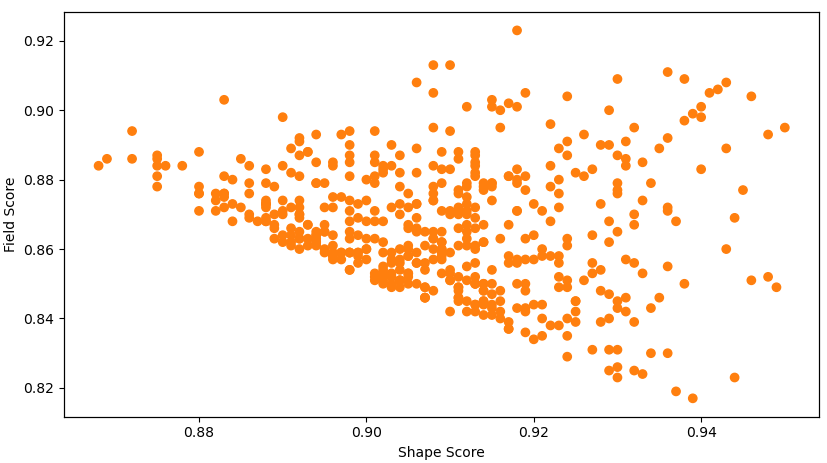
图8. Spark设计的500个大环分子与起点分子之间的形状与场相似性分布
Spark生成的大环分子的形状与场相似性综合打分(score)也相当高,分布在0.876-0.925之间,各个分值段的具体分布如下图9所示。显然,比起Yu等人1对MacroTransformer生成的大环分子采用0.65作为Xed场点相似性的截断值进行过滤而言,本次实验得到大环分子具有更高的Xed场点相似性打分。
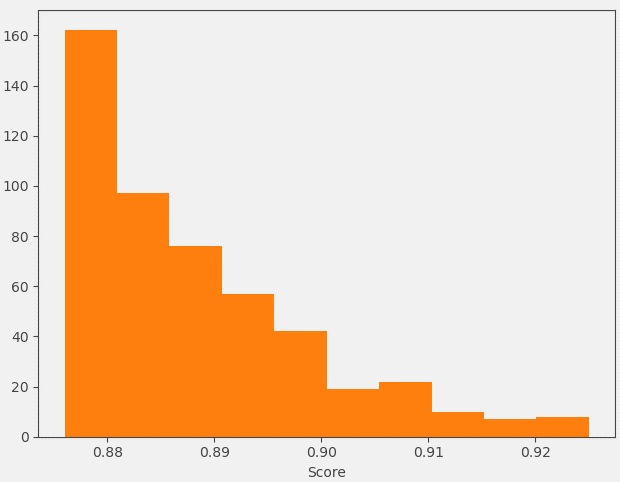
图9. Spark设计的500个大环分子与起点分子之间的形状与场相似性综合打分值(score)分布
重要的是QR-6401(化合物23)以高分的方式被SPARK直接设计出来,而在Yu等人1的研究中,深度学习MacroTransformer大环化计算并没有生成该化合物,是在之后的先导化合物优化工作中通过结构修饰而发现的。观察Spark的结果,如图10所示,QR-6401的排名靠前:按化学类型排名(Cluster Rank)第7,按化合物自身的打分排名14。其形状相似性、场相似性与总体打分(score)分别为0.938,0.897与0.918,其中打分值的取值范围为0-1:0代表没有相似性,1代表完美一致。
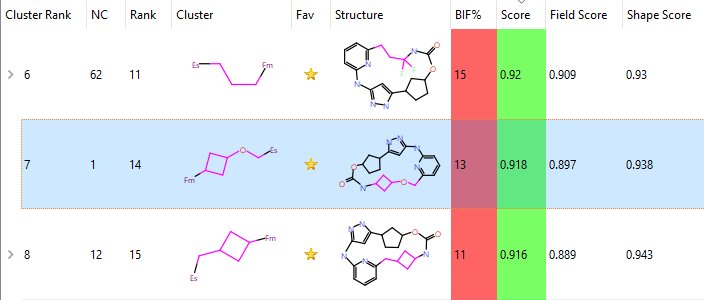
图10. QR-6401(23)的打分与排序
将Spark生成的QR-6401与作为模板的起点分子叠合观察,如图11所示,可以发现QR-6401与13的公共结构部分基本完全重合在一起,这决定了QR-6401与13具有高度的形状与场相似性。其中QR-6401的环丁烷片段与13的碳酸酰胺末端的异丙基重合非常好,环丁烷完美地模拟了异丙基的形状。如果你浏览全部的500个Spark结果,会发现很多连接臂存在环或侧链以模拟13的末端异丙基。
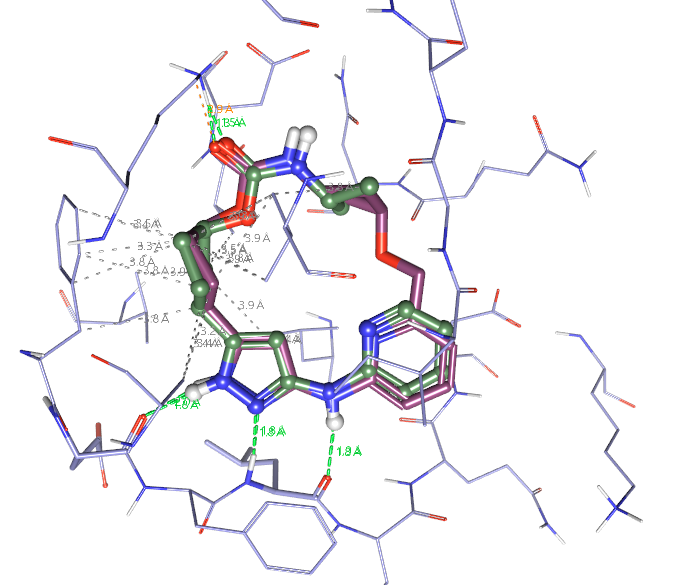
图11. QR-6401与起点分子的叠合比较。棍棒模型分子绿色为化合物13,棕色为QR-6401。
除了QR-6401之外,Yu等人1报道的部分5原子长度的连接臂也被Spark大环化工作流生成出来。
比如化合物14,如图12所示,在500个大环化合物里排名第286,而排名30的化合物因为有个取代基与13的异丙基重合导致相似性打分更高,这直接导致五碳连接臂在全部化学类型里排名17。
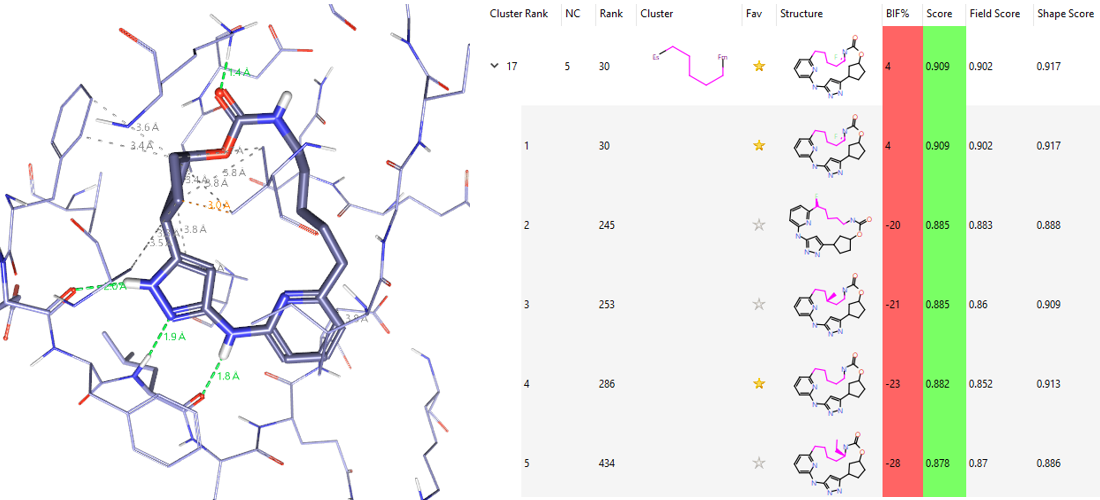
图12. 五碳连接臂。左:化合物14,Rank#286;右:五碳连接臂的排名以及典型化合物的打分与排名。
化合物15所代表的醚类连接臂在大环里排名28,如图13所示,因为引入不同的侧链而导致打分值差异导致在排名上有很大的差异。
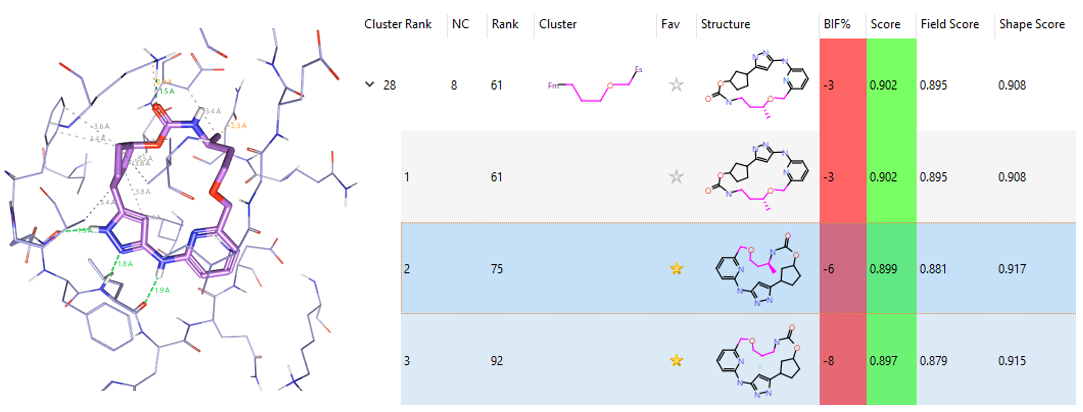
图13. 醚类连接臂。左:Rank#75;右:其它代表性化合物及排名。
除了五原子长度的连接臂之外,SPARK还生成多个3、4原子长度的连接臂,如图14所示的脂肪烃连接臂与醚连接臂。

图14. 部分3、4原子长度的连接臂示例
总的来说,Spark的大环化实验不但直接设计出了高活性、代谢稳定的QR-6401,而且设计出了其它高活性的5原子长度连接臂的大环化合物,还有许多结构多样、打分高的连接臂,相信这些连接臂中包含很多值得进一步研究的化合物。
此外,Spark的大环化计算实验直接设计出QR-6401,而在MacroTransformer实验中并没有生成。在Yu等人1的研究中,采用深度学习MacroTransformer发现了大环先导化合物,然后在先导化合物的优化工作中发现了活性、选择性与DMPK性质俱佳的QR-6401。而在Spark的大环化结果中,QR-6401在500个分子中排名14,在全部类别化合物中排名第7,相信有经验的化学家可以很快从Spark设计相中这个化合物。
结论
本文以一个“线型”的CDK2抑制剂为起点分子,经过分子对接预测结合模式,并用SPARK大环化工作流成功地设计出文献报道的大环CDK2抑制剂QR-6401以及其它化学类型的五原子长度连接臂的大环化合物,除此之外,在高分的化合物中,还包含许多打分高但没有被文献报道的新类型化合物。
补充
从共晶结构出发进行大环化
今天(2023-2-27)发现8H6T已经可以从PDB上下载,因此直接用Flare进行结构准备,然后用SPARK采用图6同样的大环化策略与图7所示的数据库进行一轮新的设计,共生成500个大环分子,包含185个不同化学类型的连接臂。QR-6401也被重现出来,不同的是,这次打分值发生变化:Field score=0.76,Shape score=0.908,Score=0.834。打分值变低,导致排名也变低,按结构类型排名80,化合物排名402,如图15所示。
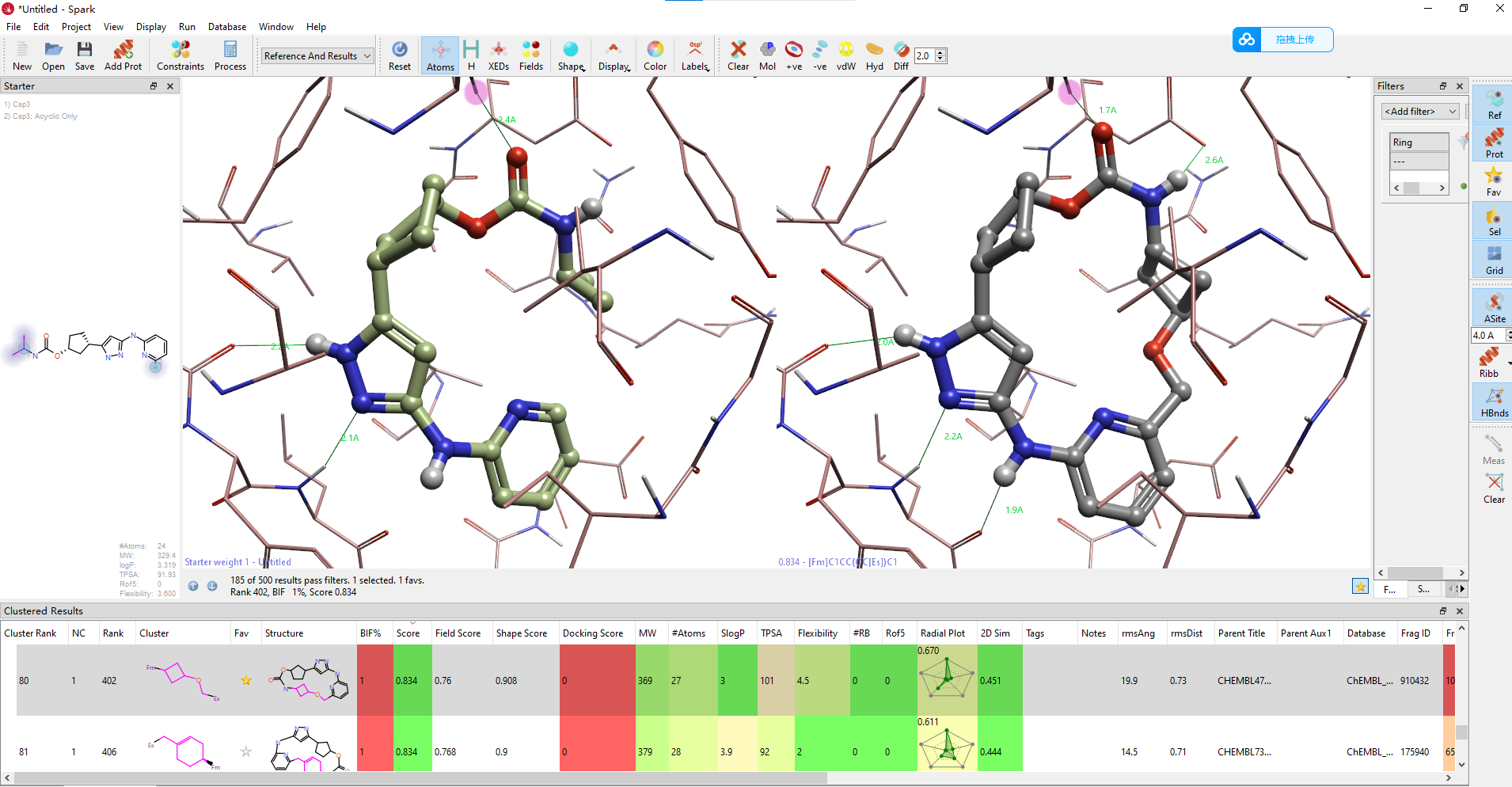
图13. 以PDB 8H6T共晶结构中的化合物13为起点分子进行大环化得到QR-6401,Cluster排名81、化合物排名402
打分值变低是因为Spark生成的QR-6401的吡唑-NH-吡啶片段更加平面化,而8H6T共晶配体的吡唑-NH-吡啶片段成八字型,这导致设计出来的分子与起点分子之间的场相似性偏低。比较13(PDB 8H6T)与19(8H6P)与CDK2结合时的活性构象可以证明这一点:13与19的吡唑-氨基的C-N两面角(如图14所示的高亮原子部分)分别为121.3°与144.2°。也就是说,大环化合物19的铰链区结合片段确实比13的更加平面化。
进一步用QM方法在B3LYP-D3BJ/6-311G(d)理论水平进行两面角扫描,如图14所示,当与CDK2结合时,13的吡唑-NH-吡啶片段能量比19的高约1.2kcal/mol。这说明,化合物13的吡唑-NH-吡啶铰链结合片段在与CDK2结合时处于一个高能的状态,大环化“摁”平了13中高能的吡唑-NH-吡啶铰链片段,使得与hinge区结合更有利而提高结合亲和力,或者说大环分子的构象张力能更低,这一点将在另一个博客里讨论。
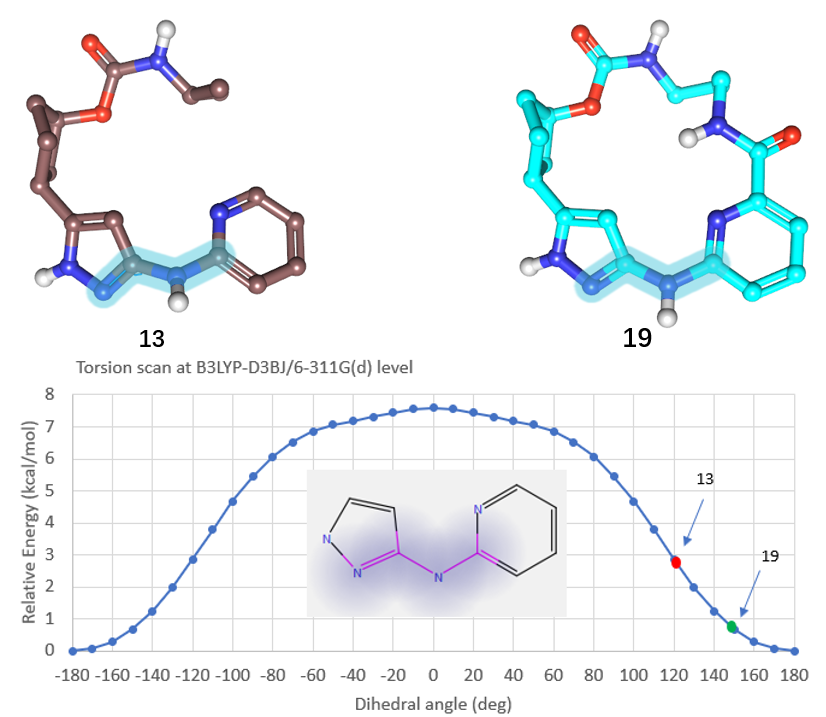
图14. 化合物13(PDB 8H6T)与19(PDB 8H6P)生物活性构象的比较,对高亮的两面角在B3LYP-D3BJ/6-311G(d)理论水平进行了扫描。
对吡唑-NH-吡啶铰链结合片段的QM计算还发现(未给出数据),当吡唑-NH-吡啶处于同一个平面上时的构象能最低,这可以解释为什么Spark大环化实验总是给出偏好于给出平面型的吡唑-NH-吡啶铰链结合片段,最终使得起点分子13使用对接构象时比使用共晶结构的构象时生成的大环化合物具有更高的打分值。
Spark命中的QR-6401连接臂哪里来?
在Spark的结果表单还包含了命中化合物的片段来源。用鼠标右键点击Rank 14(QR-6401)结果,选择“Show parent structure for select result”可以呈现Rank 14连接臂的来源母体化合物,结果如图15所示。我们可以看到,这本次大环计算中,甲氧基环丁烷片段来源于ChEMBL_rare片段库的CHEMBL4750699这个化合物,该片段在ChEMBL_rare不同的化合物中出现过10次。

图15. QR-6401的连接臂甲氧基环丁烷片段母体来自ChEMBL_rare数据库。
尽管QR-6401的连接臂在ChEMBL里属于罕见片段,仅在10个不同的化合物里出现过,但是可能高频出现于Spark其它的数据库里。如图16所示,连接臂还出现在专利数据库SureChEMBL_verycommon、商业数据库VeryCommon以及我自用的Enamine化合物库。其中,在专利数据库SureChEMBL_verycommon里不同的1351个化合物里出现过,在商业化合物库VeryCommon里不同的1227个化合物里出现过,在我自己用的Enamine(2022年版)里仅出现过两次。

图16. QR-6401的连接臂甲氧基环丁烷片段在Spark数据库里的分布与频次。
Spark的配体表单还可调出命中该片段的过程:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | Initial scores ============== Fast score 0.902, fragment 910432 from database ChEMBL_rare (version 7, created 2023-01-17 19:58:50 UTC). molecule Untitled renamed to [Fm]C1CC(OC[Es])C1 Molecule minimized, iters 786 of 5000, gradient 0.096 kcal/A (bailout 0.100). Heavy atom RMSD to starting structure 0.43A. Start energy 461.85 kcal/mol. Final energy 108.45 kcal/mol. Fields recalculated. Reference info ============== Reference "13-alt", weight 1.00 Final scores ============ Molecule realigned to starter and optimized for best score. Similarity: 0.918 Field Similarity: 0.897 Raw Field Score: -125.501 Penalised Field Score: -125.501 Shape Similarity: 0.938 Raw Shape Score: 204.166 Penalised Shape Score: 204.166 Excluded Volume Clash Penalty: 0.000 Field Value Penalty: 0.000 |
记录清楚的显示,Spark首先从创建于2023-01-17 19:58:50 UTC的CheMBL_rare数据库(第7版)中发现了这个片段,ID为910432,初始的打分为0.902,经过优化之后打分为0.918,场相似性=0.897,形状相似性=0.938。
手动旋转碳酸酰胺的C-N键是如何实现的
化合物13对接之后得到的是反式酰胺的构象,为了获得正确的pose,需要对酰胺键进行旋转,在Flare可以用配体的编辑器来完成:鼠标右键点击化合物13,选择Edit,在编辑状态下将鼠标悬停在酰胺的C-N键上方,按住左键拖拉可以旋转高亮的单键。更详细的介绍与演示请参见:
用AutoT&T的自动裁剪、移植策略对PF-07104091进行优化设计获得化合物13
Automatic Tailoring and Transplanting (AutoT&T v2.0)软件9,10是一种基于靶标结构的药物分子设计方法,可以实现先导化合物的从头设计以及结构优化等一系列功能。它可以对指定先导化合物的化学结构进行自动剪裁和移植,选取源自于虚拟筛选结果的参考分子库中与靶标蛋白结合较好的片段,嫁接到先导化合物上需要进行改造的位点,从而得到可能与靶标蛋白具有更好结合能力的分子结构。
用Flare将之前整理的CDK2共晶结构按Cα原子最小二乘法叠合到PDB 4KFU的Chain A,并提取配体作为AutoT&T V2的参考分子;并将PF-07104091对接到PDB 4KFU共晶配体的结合位点,将得到的结合模式作为AutoT&T V2的先导分子。如图17所示,用AutoT&T V2的LinkLeadOpt对PF-07104091的高亮部分进行优化设计。具体的操作过程,请参见我前文的描述11。
图17. 以PF-07104091为起点分子,对高亮部分进行替换设计
结果表明,化合物13直接被AutoT&T设计出来。进一步分析发现,AutoT&T将PDB 1GIH共晶配体(图18左)的吡啶裁剪下来,移植到PF-07104091上,并将图17的高亮部分进行了替换。1GIH共晶配体与PF-07104091具有非常相似的形状与药效团特征,如图18所示,它们与CDK2结合时的相互作用模式非常一致。
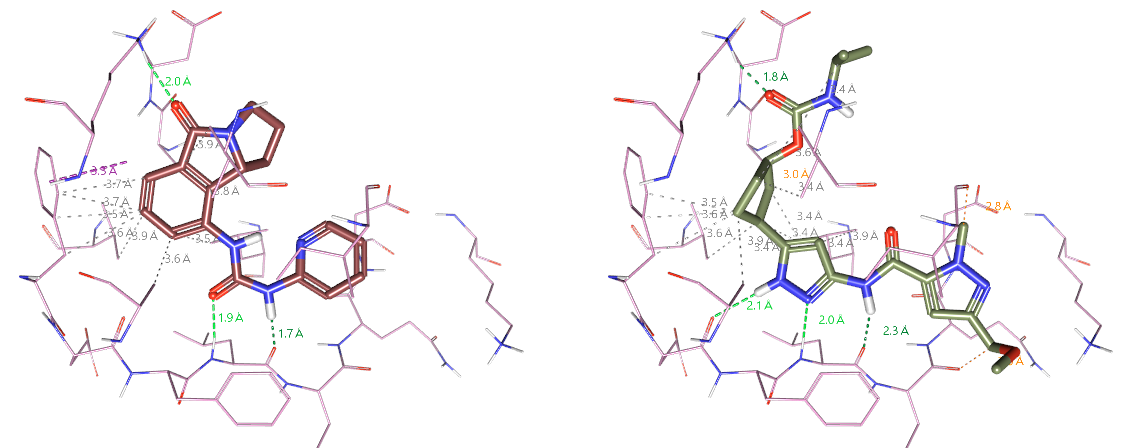
图18. 左:PDB 1GIH配体的结合模式;右:预测的PF-07104091结合模式
将PDB 1GIH的共晶配体与预测的PF-07104091结合模式进行叠合比较,如图19,可以发现PDB 1GIH配体的2-胺基吡啶片段与PF-07104091的氨基甲酰吡唑环在同一平面内完美重合,这进一步确认了AutoT&T的结果是可靠的。
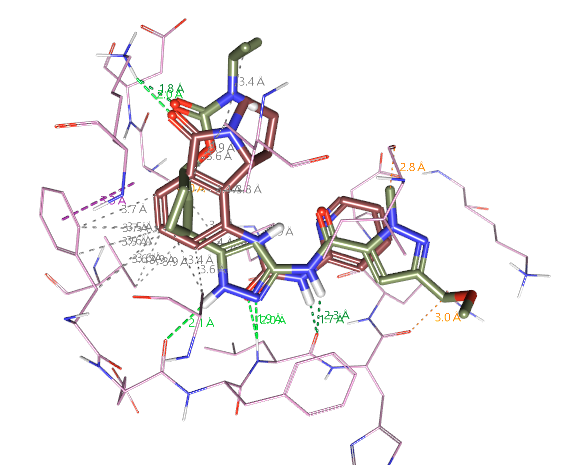
图19. PDB 1GIH配体与预测的PF-07104091结合模式的叠合比较
总的来说,通过AutoT&T对现有的共晶结构数据进行简单的探索,可以直接、理性地设计出化合物13。
Spark与AI结构生成是互补的
Spark的成功显然依赖于其庞大的片段数据库与试剂数据库。如果理想的片段或试剂没有出现在数据库里,则没有机会生成包含该片段的化合物。好在Spark提供了生成片段数据的功能12,13,你可以对AI生成的分子(包括片段)进行碎片化得到新的Spark可搜索数据库。
接下来可以作什么?
文献
- Yu, Y.; Huang, J.; He, H.; Han, J.; Ye, G.; Xu, T.; Sun, X.; Chen, X.; Ren, X.; Li, C.; et al. Accelerated Discovery of Macrocyclic CDK2 Inhibitor QR-6401 by Generative Models and Structure-Based Drug Design. ACS Med. Chem. Lett. 2023. https://doi.org/10.1021/acsmedchemlett.2c00515.
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46 (2), 665−676.
- Cheeseright, T. J.; Mackey, M. D.; Melville, J. L.; Vinter, J. G. FieldScreen: Virtual Screening Using Molecular Fields. Application to the DUD Data Set. J. Chem. Inf. Model. 2008, 48 (11), 2108−2117.
- https://www.cresset-group.com/software/spark
- https://www.cresset-group.com/software/flare
- Tosco, P.; Mackey, M. Lessons and Successes in the Use of Molecular Fields. In Comprehensive Medicinal Chemistry III; Elsevier, 2017; pp 253–296.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. J Comput Aided Mol Des 1994, 8 (6), 653–668.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. II. Organic Complexes. J Comput Aided Mol Des 1996, 10 (5), 417–426.
- Li, Y.; Zhao, Y.; Liu, Z.; Wang, R. Automatic Tailoring and Transplanting: A Practical Method That Makes Virtual Screening More Useful. J. Chem. Inf. Model. 2011, 51 (6), 1474–1491. DOI:10.1021/ci200036m
- Li, Y.; Zhao, Z.; Liu, Z.; Su, M.; Wang, R. AutoT&T v.2: An Efficient and Versatile Tool for Lead Structure Generation and Optimization. J. Chem. Inf. Model. 2016, 56 (2), 435–453.DOI:10.1021/acs.jcim.5b00691
- 肖高铿. 墨灵格的博客. AutoT&T算例——DDR1抑制剂从苗头化合物到先导化合物的发现.2021-11-29. http://blog.molcalx.com.cn/2021/11/29/ddr1-from-hit-to-lead.html
- 分子碎片化创建自己的可搜索数据库. 墨灵格的博客. 2021-04-06. http://blog.molcalx.com.cn/2021/04/06/spark-molecule-fragmentation.html
- 如何导入试剂创建自己的可搜索数据库. 墨灵格的博客. 2021-03-26. http://blog.molcalx.com.cn/2021/03/26/reagent-import.html


