
摘要:本教程以COX-2抑制剂为例,演示了如何用Forge建立3D-QSAR模型的流程:1)导入参比化合物;2)导入训练集化合物与3)测试集化合物;4)自动构象搜索、分子叠合,PLS分析,生成模型与模型验证。通过本练习,您会掌握Forge的使用方法、并实现分子结构导入,构象搜索以及分子叠合与3D-QSAR建模、分析与靶点预测。
作者:肖高铿
联系:gkxiao@molcalx.com
一. 背景
本教程以COX-2抑制剂为例讲解如何用Forge建立COX-2抑制剂的3D-QSAR模型。
建模前准备工作:
1)训练集化合物:training.sdf
2) 测试集化合物:test.sdf
3) 确认参比化合物:reference.sdf, 一般为活性最高的化合物的一个可能活性构象(另有教程介绍如何预测活性构象)
二. 操作步骤
- 新建项目,使用项目模板开始3D-QSAR建模
- 读入参比化合物
- 读入训练集化合物
- 读入测试集化合物
- 读入靶点蛋白
- 可选步骤:读入待预测活性化合物
- 设定参比化合物的场
- 开始建模:构象搜索、分子叠合、3D-QSAR模型生成多任务一键自动完成
- 模型的选择与分析
File > New Project 或者点击菜单栏New按钮建立一个新的项目
弹出New Project对话框,选择一个项目模板,点击“Build Activity Model”使用该模板开始3D-QSAR建模, 如图1所示。
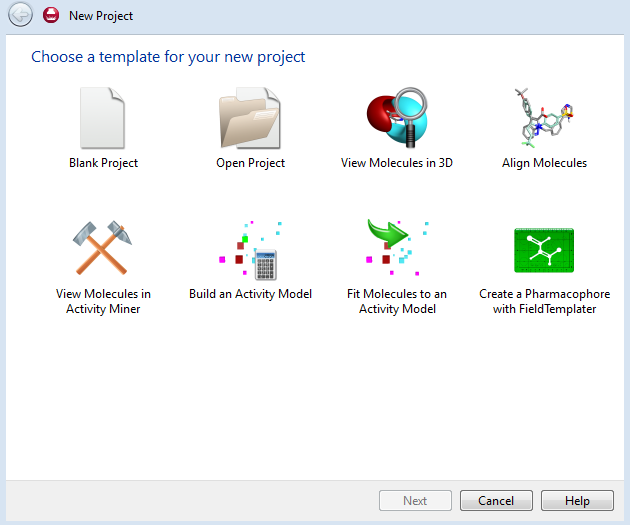
图1. Forge的项目模板
Forge提供了多种QSAR建模方法,适用于不同的目的,本文要建立3D-QSAR模型,所以选择Field QSAR Model,如图2所示。
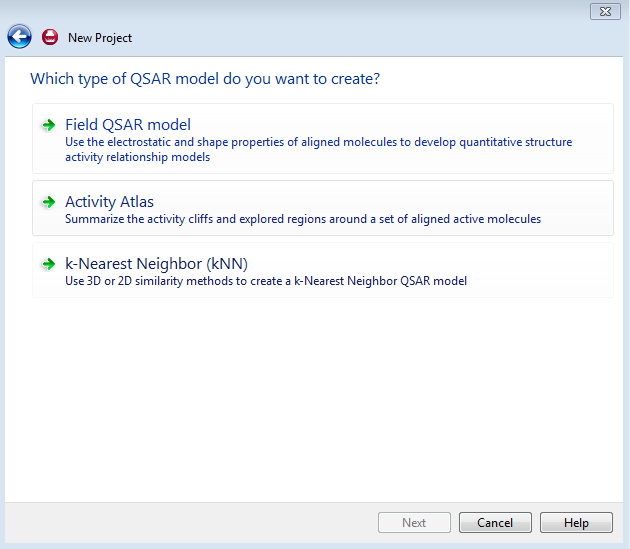
图2. Forge的QSAR模型有三种:Field QSAR Model(3D-QSAR),Activity Atlas与kNN。
Forge可以根据自己的数据集是否预叠合进一步进行选择:你可以用第三方软件比如分子对接预先叠合好化合物,也可以让Forge进行构象搜索、再进行化合物叠合。
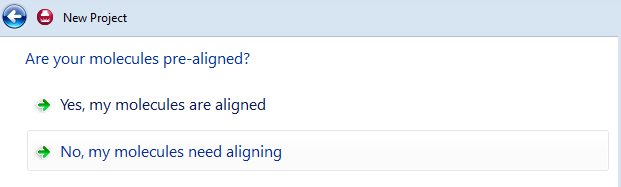
图3. 可以使用预先叠合好的分子结构也可以让Forge对化合物进行构象搜索、分子叠合
如果你的化合物还没有进行叠合过、则需要选择:No, My Molecules need aligning.
Forge会进一步提问:你的数据是否已经分组为训练集与测试集。本教程我们以及预先分成两组,因此选择Yes,I have seperate training and test sets.
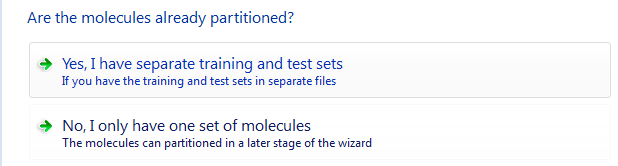
图4.Forge的3D-QSAR建模需要对数据进行分组:训练集与测试集
Forge接着提示导入参比化合物(见图5),注意:参比化合物一定是个化合物的3D构象,同一个化合物不同的构象建立的3D-QSAR模型很可能会非常不同,最合理的构象是活性构象。活性构象可以1)从配体-受体复合物结构提取,2)也可以用Forge的FieldTemplater模块来预测化合物的活性构象;3)还可以用Forge对参比化合物进行构象搜索,取低能构象集,每个构象分别作为参比化合物建立3D-QSAR模型,然后从多个3D-QSAR模型择优使用。
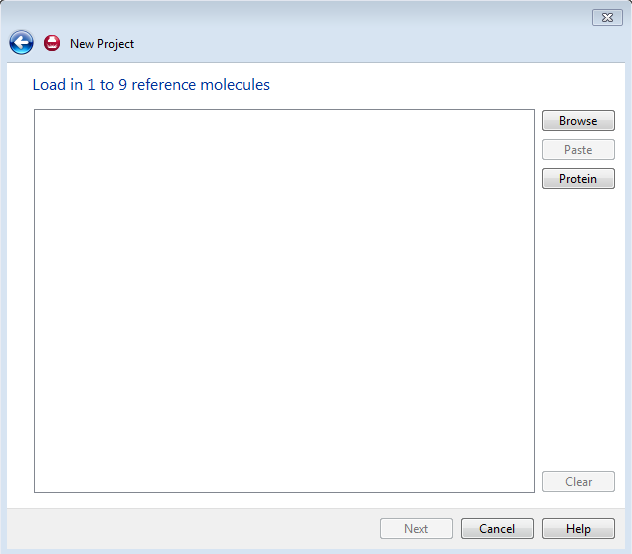
图5. 导入参比化合物
点Broser导入化合物,Forge接着会是否使用Forge来定义质子化状态还是用输入文件的质子化状态,这里我们直接使用化合物输入文件的质子化状态就可以,见图6。
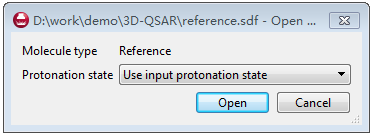
图6. 参比化合物的质子化状态设定
点击Next按钮进入下一步读入训练集化合物。
基本流程与上一步读入参比化合物一致,根据情况选择读入模式。本教程中每个化后已经生成好3D结构,但构象没有搜索、也没有进行过叠合,选择autodetect就可以,见图7。
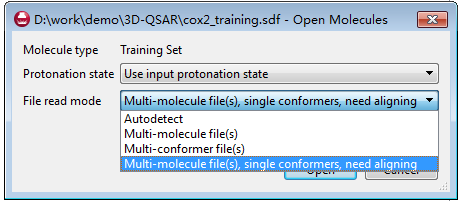
图7. 训练集化合物的读入模式
点击Next按钮,进入下一步,进行活性数据设定,见图8。
图8. 活性数据的设定:需要预先在SDF文件将化合物的活性数据体现在结构文件里。
同“读入训练集化合物”操作步奏。
如果参比化合物从配体-受体复合物结构提取出来的活性构象,还可以读入该蛋白结构以便叠合时考虑到蛋白的“bump”影响。
对场点进行编辑可以获得更好的计算结果,编辑完毕点Finish结束进入下一步骤。
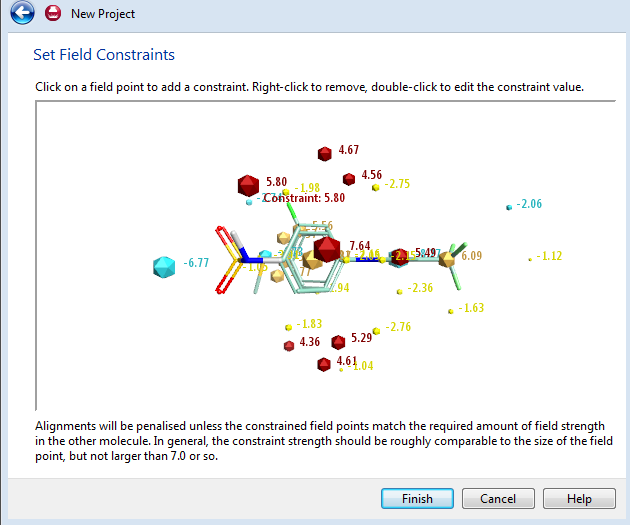
图9. 可以根据您对项目的了解,对参比化合物的场点进行编辑:删除无用的场点、约束长点大小等。
Forge processiong对话框,点击Start开始构象搜索、分子叠合与3D-QSAR建模。,也可以分别代开构象搜索与分子叠合模块进行个性设定,最后点击Start按钮开始作业。
其中构象搜索的参数可以参考:http://blog.molcalx.com.cn/2016/06/04/torch-tutorial-conformation-hunting.html
其中分子叠合可以参考:http://blog.molcalx.com.cn/2016/06/04/torch-tutorial-align.html
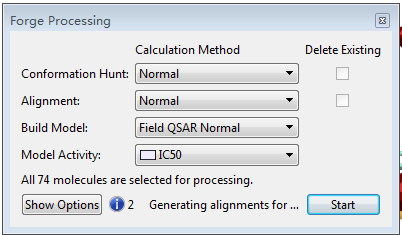
图10. Forge processiong对话框,点击Start开始构象搜索、分子叠合与3D-QSAR建模。
上一步结束之后会自动生成统计学参数以评估模型质量、还可以观察分子的形状与静电场对活性的影响。
第一步:确认主成分数
不同主成分数的模型不仅Q2与R2不同(图11),而且预测误差RMSE也不同(图12)。在Forge里,用鼠标点击不同的位置,会选择不同的主成分数给出统计学结果、建立不同的模型。
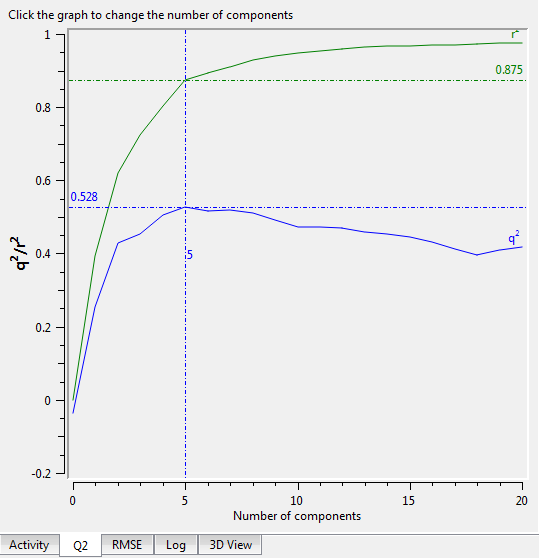
图11. PLS主成分数 VS Q2(蓝色)与R2(绿色)
我们会发现R2是主成分数的单调上升函数,而Q2不是主成分数的单调函数。Q2的计算方法如下:
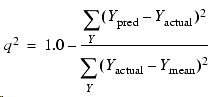
对于一个新的化合物,如果不采用任何计算方法也可以用训练集化合物活性的平均值做预测值,这时Q2计算公式右边里的分子与分母一样,则Q2=0。也就是说,不采用任何计算方法建立的模型的Q2为0。而一个完美模型的预测值会等于实验值,这时Q2计算公式右边里的分子为0,则Q2=1。介于不用模型与完美模型之间的Q2值为0-1之间。因此,Q2实际反映了:比起不用计算方法,该计算模型值不值得做。一般认为,可以接受的模型的Q2应当大于0.5。
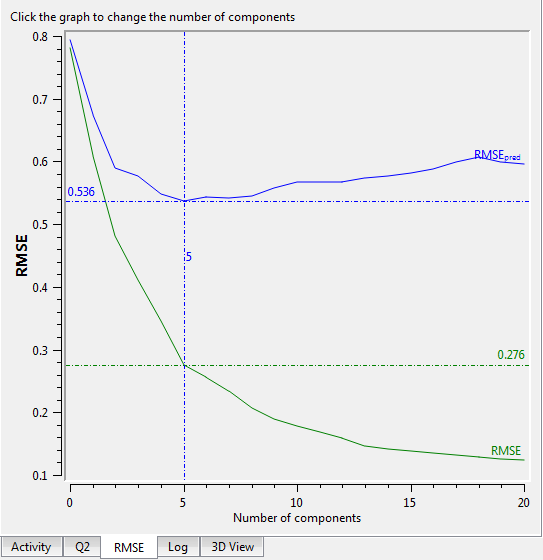
图12. PLS主成分数 VS RMSE。其中RMSE指整个训练集建的模型的RMSE,而RMSEpred指交叉验证RMSE。
如图12所示,非交叉验证的RMSE是主成分数的单调下降函数,而交叉验证的RMSEpred先下降或上升。这提示:如果只看RMSE值,而不考察RMSEpred很可能过拟合,主成分数可能取太大了。
主成分数的取值应该考虑几个方面:1)RMSE与RMSEpred越小越好。如图12所示,因为主成分数等于5时RMSEpred最低,所以主成分数最大取5。2)还要考察Q2,取Q2最高的那个主成分数。在本例,当主成分数为5时,Q2最高。当1)与2)有冲突时,取主成分数小的那个。3)依次降1个主成分数,看主成分数高的模型Q2是否比低的那个Q2高出5%以上,否则用主成分数小的模型,这么做的主要目的是保证信噪比要足够好。当然,这些都是要有前提:训练集数据不得冗余。也就是相似化合物的活性不能相似,不相似化合物的活性相似。因为我们在训练模型时采用Leave-one-out交叉验证(LOO CV),如果数据冗余,抽掉一个化合物之后的训练集里还有一个跟它结构相似活性相似的分子,就像考试带小抄一样,这个模型的预测性能就会被高估。
第二步:观察模型
Forge的Field-based 3D-QSAR模型不仅是预测性模型,而且还是解释性模型。点击3D View可以考察静电场、立体场对活性的影响(图13)。更多的信息需要亲自练习。
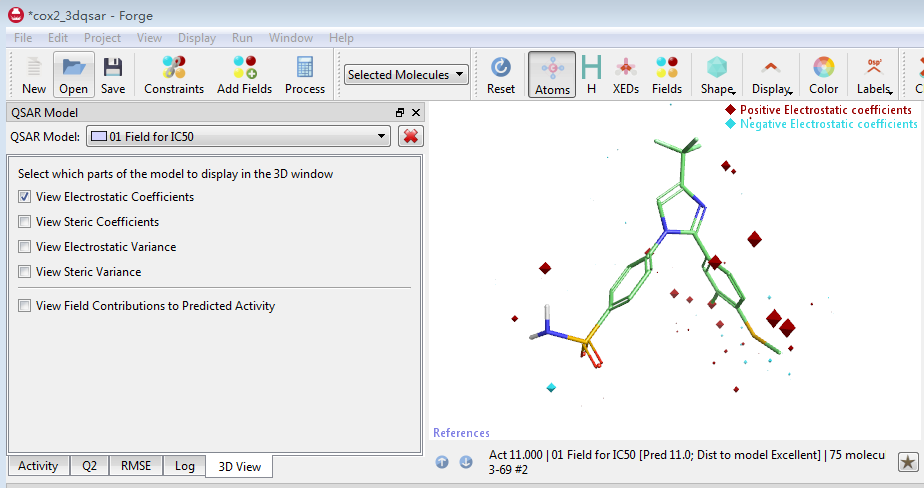
图13. 3D-QSAR的等值图可以分析静电场、立体场对活性的影响
三. 注意事项
1. 参比化合物的构象很重要
可以用药效团方法、分子对接方法预测化合物的可能活性构象,或者用构象分析方法搜索可能的活性构像:每个构象建立一个3D-QSAR模型。模型越多,命中的概率越大。
2. 参比的化合物只需提供一个构象并且不行进构象搜索
需要对训练集化合物与测试集化合物进行构象搜索、并且叠合到参比化合物上;参比化合物采用生物活性构象,没有进行构象搜索。
3. 输入文件要求
推荐采用SDF格式,并将活性值以IC50或pIC50的方式表达,包含在SDF文件里。
四. 接下来可以做什么?
1,为参比化合物生成多个构象,每个构象依次建模,获得最可能的3D-QSAR模型;
2,用3D-QSAR模型指导结构改造;
3,用3D-QSAR用来预测新化合物的活性。
五. 相关文献
- Giuseppe, F.; Emanuele, A.; Maria, D.; Agostino, M.; Loredana, S.; Giuseppe, R.; Orazio, P.; Valeria, P.; Antonio, R. Identification of Potentially Potent Heme Oxygenase‐1 Inhibitors through 3D‐QSAR Coupled to Scaffold Hopping Analysis. ChemMedChem 2018.
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistarà, V.; Pittalà, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR Statistical Model-Scaffold Hopping Analysis for the Identification of Potentially Potent and Selective Sigma-2 Receptor Ligands. Eur. J. Med. Chem. 2017, 139, 884–891.
- Alam, S.; Khan, F. 3D-QSAR Studies on Maslinic Acid Analogs for Anticancer Activity against Breast Cancer Cell Line MCF-7. Sci. Rep. 2017, 7:6019.
六. 试用
软件下载:http://www.cresset-group.com/try-a-free-demo
信息提交:


