
Flare™V7.0发布——增强的分子对接、更好的QSAR建模与更精确的FEP与MD结果
摘要:Flare是Cresset基于配体和基于结构的建模平台,本文介绍了最新版本V7全新的、增强的科学和方法,包括用大正则非平衡候选蒙特卡罗(GCNCMC)水合模式增强采样的分子动力学模拟与FEP计算、QM半经验计算、系综共价对接方法以及构建预测性QSAR模型的共识模型等等。
Flare V7简介
Cresset基于配体和基于结构的建模平台的新版本7包含了全新的、增强的科学和方法,包括用大正则非平衡候选蒙特卡罗(GCNCMC)1水合模式增强采样的分子动力学模拟(Molecular Dynamics)与自由能微扰(Free Energy Perturbation,FEP)计算,QM半经验计算,新增系综共价对接方法,以及用于构建预测QSAR模型的共识模型(Consensus models )。
本次发布还扩展了可用工具的选择,以解决Flare FEP实验问题、分析动力学轨迹、为动力学和FEP模拟创建配体自定义参数,并为进一步研究准备配体结构。
新的并行处理以及计算窗口可以在同一Flare项目中启动、运行和监控多个作业,使平台更加高效、友好协作、使用更简单。
更高效的水分子采样能力并提高了Flare FEP预测性能
在Flare V7中可使用GCNCMC进行更高效的水分子取样,用于分子动力学和FEP实验。该方法将传统的正则蒙特卡罗(GCMC)2与非平衡候选蒙特卡罗(NCMC)相结合,以渐进、非平衡的方式进行水的插入和删除,改善了蛋白活性位点水合模式的采样。
在Flare FEP计算的平衡阶段使用GCNCMC可以显著提高该方法的预测能力,特别是在蛋白-配体复合物中,其中活性位点具有封闭的口袋,通常难以发生水合作用的时候(图1)。

图1. 在Flare FEP计算平衡阶段将配体A(从PDB:2QBP,PTP1B Ki=4nM3)转化为配体B(PTP1B Ki=2000nM3)时使用GCNCMC显著提高了FEP预测的准确性。
增强的FEP分析工具
子图分析是活性图的一个新功能,它进一步扩展了Flare用于分析FEP基准实验结果的工具选择。在Flare FEP项目开始时运行,以确保系统设置正确,通过比较活性图中每个配体的实验与预测ΔG,确认该方法对所研究的蛋白靶标/配体系列的预测能力(图2)。

图2. Flare FEP项目的活性绘图。
在基于真实药物发现数据运行的Flare FEP项目的微扰网络中,经常观察到一些粒度——高度同源、高度互联的化合物小组。这些组中分子的相对ΔΔG预测通常精确可靠的。然而,如果与网络其他部分的连接有问题,或者如果网络包含了预测精度较低的化合物,则整个数据集的统计数据会受到影响,导致对表现良好组中化合物的预测误差被高估,而相对ΔΔG预测却完全可信。
在这种情况下,子图分析可以用来识别具有较低内部误差统计的化合物簇,其中预测更可靠,并突出导致FEP项目中的统计误差大、有问题的转化。如图3中项目所示,发现了两簇化合物。在簇#1(紫色)中化合物的预测不太精确,如活性图中所示具有大的误差条,因为许多连接受到一定程度的滞后影响。在簇#2(绿色)中分子的预测反而更精确(误差条更小)。因此,我们可以高度信任#2中分子的相对ΔΔG预测,但不太信任#1中、或#1和#2之间分子的预测,因为这些连接也显示出滞后(橙色椭圆)。

图3. Flare V7中的子图分析有助于识别具有较低内部误差统计的化合物聚类,其中预测更可靠,并能够识别出在FEP项目中导致较大统计误差的有问题的转化。
新增半经验方法进行更快速的QM计算
新增的基于GFN2-xTB45实现的半经验紧束缚方法进一步扩展了Flare执行量子力学(QM)计算的可选项。推荐将该方法用于大型配体的几何优化和单点能计算,其精度适中。此外,得益于Flare的灵活实现,可以通过运行快速的半经验最小化,然后再在DFT级别进行能量优化来提高QM工作流的计算效率(图4)。

图4. 大环内酯类阿奇霉素在半经验理论水平上的几何优化,然后在真空或溶剂中进行DFT水平的单点能量优化,只需几分钟就可以在笔记本电脑上完成,从而为更大的配体实现计算高效的工作流。青色表面 = QM电子密度。
pyflare用户还可以使用新增的“QM.py”脚本从命令行访问计算效率高的QM计算。
新增分析工具增强分子动力学模拟
对于分子动力学实验,可以在平衡阶段或整个模拟过程中激活GCNCMC选项。这能够在模拟中增强对配体周围蛋白活性位点区域内水合模式的采样,并且通常导致对结合型配体构象采样的增加(图5)。高级选项可对GCNCMC球体大小和移动频率进行微调。

图5.在分子动力学模拟中,GCNCMC选项可以仅在平衡阶段激活,也可以在整个模拟过程中激活。
新的“导出轨迹(Export Trajectory)”按钮(图6-左)可用于导出整个动力学轨迹,或仅导出所需的帧范围,为“修剪”轨迹以便仅包括感兴趣范围内的帧提供了高效的工作流。然后,可以通过按下“打开轨迹(Open Trajectory)”按钮(图6–右),将修剪后的轨迹导入Flare,并将其与导出的蛋白相关联。

图6. 左:新的“导出轨迹(export trajectory)”按钮可用于导出整个动力学轨迹,或仅导出感兴趣范围内的帧。右:然后可以按“打开轨迹(open trajectory)”按钮将导出的轨迹导入回Flare中。
动力学轨迹的聚类也得到了增强,能够仅对选定范围的帧执行分析。
新增方法和增强方法用来精确自定义扭转力场参数的计算
精确的扭转力场参数对于可靠地预测小分子配体和辅因子的热力学性质至关重要。在Flare V7中,我们进一步扩展了创建可选自定义扭转参数的算法选择,以支持分子动力学和Flare FEP计算,包括新增的混合DFT//GFN2-xTB方法(图7)。
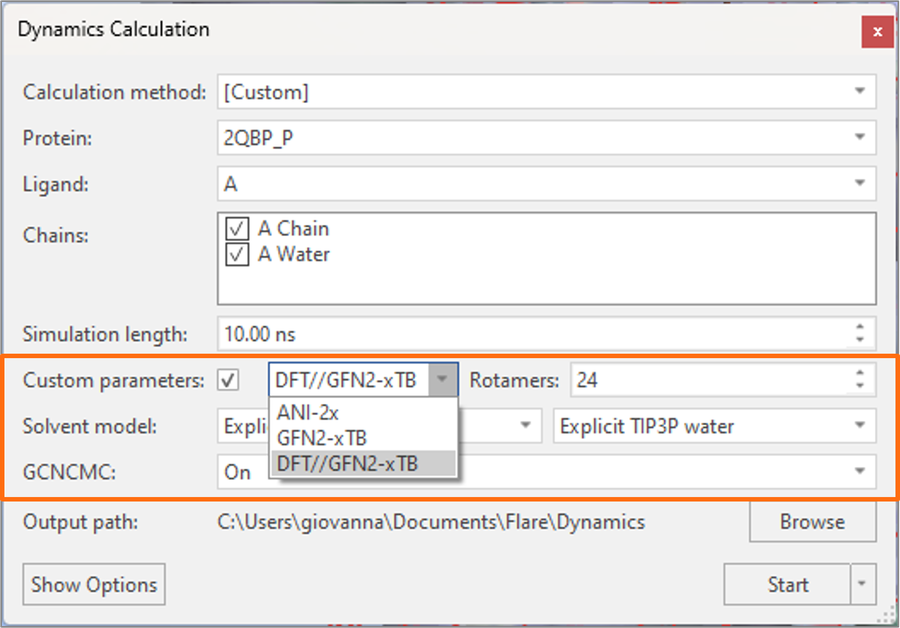
图7. Flare V7提供了一个扩展的算法选择,用于为Open力场创建自定义扭转参数
强烈建议所有小分子(中性和带电6)使用该方法,在使用GFN2-xTB紧密结合半经验方法获得的几何结构上计算B3LYP-D3BJ/DZVP理论水平的DFT单点能量。通过这种方式,由于快速的半经验几何优化,可以获得良好的计算性能,同时采用从头计算方法进行高质量的能量计算。
现有的ANI-2X7和GFN2-xTB5方法也已使用增强的碎片化算法进行了更新。
用增强的QSAR功能,构建预测性模型
在Flare现有稳健且经过充分验证的机器学习(ML)方法中新增了“共识(Consensus)”回归和分类模型(图8)。运行共识回归模型(图8-左)将导致Flare运行高斯过程、多层感知器、随机森林和支持向量机模型,并将化合物的活性预测为每个单独模型预测的平均值。共识分类(图8右)将分子的类别预测为多层感知器、随机森林分类模型预测概率总和最高的类别。
这创造了一个“共识”结果,使你能够在对配体进行优先性排序的时候,优先考虑所有回归或分类模型都认为是好主意的那些配体。

图8. 共识回归(左)和分类(右)模型使您能够对配体进行优先性排序,优先考虑所有机器学习回归或分类模型一致认为可能是好主意的配体。
在Flare V7中,可以选用不同的RDKit指纹8(图9-左)无缝构建机器学习回归和分类模型。此外,几种常用的物理化学描述符9可以从RDKit导入到配体表单(图9-右),并用作静电和形状的Cresset 3D描述符的替代品,用于构建活性和ADMET性质的预测性QSAR模型。

图9. 在Flare中,RDKit指纹和物理化学描述符可以用于建立活性和ADMET性质的预测性QSAR模型。
最后,高斯过程回归模型现在可以计算每个预测值的标准偏差值,使您能够在进行化合物合成和测试之前对Flare所做预测的可靠性获得信心。
更多的分子对接与打分选项
在Flare V7中新增的“系综共价(Ensemble covalent)”方法融合了两种流行的对接方法:1)共价对接,为共价结合的抑制剂产生合理的结合模式;以及2)系综对接,通过在单个对接实验中包括相同蛋白质多个可选的活性位点构象来考虑活性位点的柔性。
与共价对接一样,待对接的配体必须携带一个共价弹头,目前Flare总共支持20多种共价弹头:请注意,如果您想要使用的共价弹头还没得到支持,可以很容易地将其添加到Flare中并与其他同事共享(详细信息请联系Cresset技术支持)。与系综对接一样,在研究中涉及的不同蛋白质构象必须在3D上叠合对齐在一起(图10)。
对接格点(grid,配体进行对接的区域)可以用已知共晶的配体位置或通过手动选择合适的蛋白质原子来轻松地定义。

图10. Flare中的计算面板使建立系综共价对接实验变得容易
只在其中一个蛋白质上定义共价残基就足够了:其他叠合上来的蛋白结构中的等效共价残基可通过Flare自动检测。或者,也可以从每个蛋白中手动挑选共价残基。
在系综共价对接实验结束时,Flare共价对接算法生成的结合模式将保存在与它们对接的蛋白相关联的配体表单中。Rank Score最低的结合模式重现正确共晶结构结合模式的可能性最高(图11)。

图11. HCV NS3/4A蛋白酶复合物共价抑制剂的系综共价对接实验结果。Narlaprevir的2D结构用于对HCV NS3/4A蛋白酶的三种不同构象(PDBs 3SV6、3LON、3LOX)进行的系综共价对接实验。Flare发现的得分最高的结合模式(右,橙色)非常接近PDB 3LON中narlaprevir的晶体学结合模式(左,绿色)。蛋白质表面根据共晶实验(左)和对接计算(右)narlaprevir的静电互补性着色(绿色=良好的静电互补;红色=静电冲突)。
在Flare V7中,还可以为您的对接实验找到新的、有用的高级选项。共价对接一个新增的高级选项使您能够通过键入SMARTS来指定用于实验的共价弹头。当待对接配体携带一个以上潜在共价弹头的情况下很有用,并且你想利用你对所研究的化学系列的了解来控制反应基团。
在Flare中,对接方法新增一个新的高级选项使您能够指定在活性位点中哪些氢键供体蛋白残基(从丝氨酸、苏氨酸、酪氨酸和赖氨酸中的每一种或全部中选择)在对接实验期间可以旋转。这提供了一种额外的方式,可以在对接实验中实现侧链柔性,并实现与蛋白质活性位点的最佳相互作用。
并发计算
通过使Flare能够并发运行多个作业,我们在本版本中显著提高了计算效率(图12)。

图12. 在Flare中并发处理多个作业
智能锁定系统可防止正在计算中的作业使用到的配体和蛋白被无意删除或修改,直到这些计算完成。此外,重新设计的计算窗口可监控所有正在运行、排队和已完成的作业,从而可以轻松访问每次计算的日志。
增强的配体准备
本版本中的配体准备(Ligand Prep)功能通过枚举互变异构体(enumerate tautomers)的新选项得到了丰富。根据Cresset规则,互变异构体枚举可以选择只列举典型的互变异构,以及在水溶液中从极有可能到极不可能的互变异异构体(图13)。
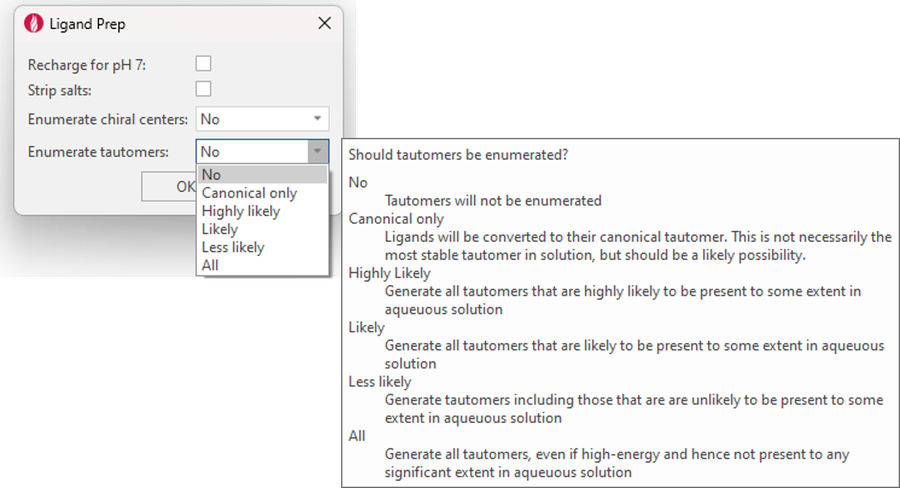
图13. 新增配体准备的互变异构体枚举选项
在蛋白准备时使用“create biological assemblies”选项
自然界中许多蛋白质的功能形式是生物学单元(biological assembly),即一个或多个单元的大分子组装:例如,有功能的血红蛋白是四条链组成的四聚体。在Flare中可以直接导入生物单元,但在有时需要一些手动干预。在Flare V7中,我们为蛋白准备添加了一个新选项,使其能够以自动化方式轻松导入在PDB中所述的生物学单元。
在图14的左侧,我们可以看到在默认条件下蛋白质准备后的PDB 6TOH(CCT36538610与BCL6结合的共晶结构,BCL6是治疗弥漫性巨大B细胞淋巴瘤的潜在治疗靶点)。在右侧,我们可以看到使用“create biological assemblies”选项导入的PDB 6TOH。BCL6的功能性二聚体结构已被自动导入,结合位点中重要的充满水的口袋清晰可见。

图14. 左:在Flare中使用默认条件导入PDB 6TOH并进行蛋白结构准备;右:使用“Create biological assembly”选项自动重新创建BCL6的功能性二聚体形式。其中活性位点的表面根据疏水性着色11。
其它增强与改进
在Flare V7中新的增强与改进还包括:
- 新增用角色(role)来组织、管理蛋白与比对表单的功能
- 增强了蛋白的建模能力:
- 增强的库枚举,具有更清晰的命名和扩展的反应定义,以及导出/导入结构过滤器的选项
- 新增Mogul扭转分析方法的接口(需要有效的CSD许可证)
- 新增Flare GUI顶部“搜索”框,用于快速定位所需的Flare功能,如功能区选项卡按钮、菜单项和可固定窗口
- 新增导出Spin/Rock视频的功能
- 交互式径向图
- 支持AND/OR逻辑操作对配体的Tags标签进行过滤
- 扩展了Hit Expander取代基的选择
- 增强了R-基团分析结果的可视化
- 情节提要中的现有场景现在可以更新
- 新增散点图中配体标题的永久注释。
(1)在编辑器了建立肽类,进行蛋白生长
(2)新增“复制/粘贴Loop”的功能,通过复制不同蛋白质结构的Loop来修复感兴趣蛋白质中的gap
(3)新增在所需位置插入残基序列的功能
(4)对于无三维坐标的蛋白质序列中残基,能够进行选择、删除、放入新链和链的分割
(5)单点突变的工作流程现在可以选择仅对突变的残基进行准备或是对整个蛋白进行准备
(6)新增蛋白和比对表单中的交互式序列标尺条,用于突出显示/选择所有蛋白质中相同位置的所有残基
文献
- O. J. Melling, M. L. Samways, Y. Ge, D. L. Mobley, J. W. Essex, Enhanced Grand Canonical Sampling of Occluded Water Sites Using Nonequilibrium Candidate Monte Carlo, J. Chem. Theory Comput. 2023, 19, 1050-1062
- M. L. Samways, H. E. Bruce Macdonald, J. W. Essex, grand: A Python Module for Grand Canonical Water Sampling in OpenMM, J. Chem. Inf. Model. 2020, 60, 10, 4436-4441
- Douglas P. Wilson, Zhao-Kui Wan, Wei-Xin Xu, Steven J. Kirincich, Bruce C. Follows, Diane Joseph-McCarthy, Kenneth Foreman, Alessandro Moretto, Junjun Wu, Min Zhu, Eva Binnun, Yan-Ling Zhang, May Tam, David V. Erbe, James Tobin, Xin Xu, Louis Leung, Adam Shilling, Steve Y. Tam, Tarek S. Mansour, and Jinbo Lee, Structure-Based Optimization of Protein Tyrosine Phosphatase 1B Inhibitors: From the Active Site to the Second Phosphotyrosine Binding Site, J. Med. Chem. 2007, 50, 19, 4681–4698
- https://xtb-docs.readthedocs.io/en/latest/setup.html
- C. Bannwarth, S. Ehlert, S. Grimme, GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comp. 2019, 15, 3, 1652-1671
- J. S. Smith, O. Isayev, A. E. Roitberg, ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost, Chem. Sci. 2017, 8, 3192-3203
- https://www.rdkit.org/docs/GettingStartedInPython.html#fingerprinting-and-molecular-similarity
- https://www.rdkit.org/docs/GettingStartedInPython.html#list-of-available-descriptors
- B. R. Bellenie, K.-M. J. Cheung, A. Varela, O. A. Pierrat, G. W. Collie, Gary M. Box, M. D. Bright, S. Gowan, A. Hayes, M. J. Rodrigues, K. N. Shetty, M. Carter, O. A. Davis, A. T. Henley, P. Innocenti, L. D. Johnson, M. Liu, S. de Klerk, Y.-Vaï Le Bihan, M. G. Lloyd, P. C. McAndrew, R. Talbot, H. L. Woodward, R. Burke, V. Kirkin, R. L. M. van Montfort, F. I. Raynaud, O. W. Rossanese, S. Hoelder, Achieving In Vivo Target Depletion through the Discovery and Optimization of Benzimidazolone BCL6 Degraders, J. Med. Chem. 2020, 63, 8, 4047–4068
- P. K. Behara, H. Jang, J. Horton, D. Dotson, S. Boothroyd, C. Cavender, V. Gapsys, T. Gokey, D. Hahn, J. Maat, O. Madin, I. Pulido, M. Thompson, J. Wagner, L. Wang, J. Chodera, D. Cole, M. Gilson, M. Shirts, C. Bayly, L.-P. Wang, D. Mobley, Benchmarking QM theory for drug-like molecules to train force fields, Poster presented at CUP XXI, March 08, 2022 – March 10, 2022, Santa Fe, New Mexico, USA
- Wimley, W. C.; White, S. H. Experimentally Determined Hydrophobicity Scale for Proteins at Membrane Interfaces. Nat. Struct. Mol. Biol. 1996, 3 (10), 842–848. https://doi.org/10.1038/nsb1096-842.
灵活的许可与商务模式
Flare具有灵活的许可选项,适合计算化学家、药物化学家和学者。


