
用多个蛋白信息来提高用单一蛋白结构分子对接虚拟筛选的性能
摘要:本文以PDE5A数据集为例,比较了使用单一apo结构的FRED分子对接虚拟筛选与使用多个holo结构的HYBRID分子对接虚拟筛选在性能上的差异。结果表明,在以AUC为代表的总体虚拟筛选性能上,HYBRID比FRED稍有优势;而在以logAUC、BEDROC、EF、ER1%为代表的早期富集能力上,HYBRID大幅优于FRED。与几个流行的分子对接软件AutoDock Vina、Surlfex、GLIDE、GOLD、FLEXX等相比,FRED与HYBRID表现出远优于其它方法的早期富集性能。
肖高铿/2020-03-01
1. 前言
FRED与HYBRID是OEDocking中两个基于分子对接的虚拟筛选应用[1]。FRED采用穷尽搜索算法(见图1),即系统性地枚举配体每个构象在特定分辨率结合位点里的平动与转动空间。在穷尽搜索的时候,将不可能的pose过滤掉,对保留下来的进行打分。搜索完毕,对打分最高的那些poes进一步进行优化(局部穷尽搜索,比全局穷尽搜索用更高的分辨率),最后将打分最高的那个pose与数据库中其它化合物进行比较、排序,完成虚拟筛选[2,3,4]。
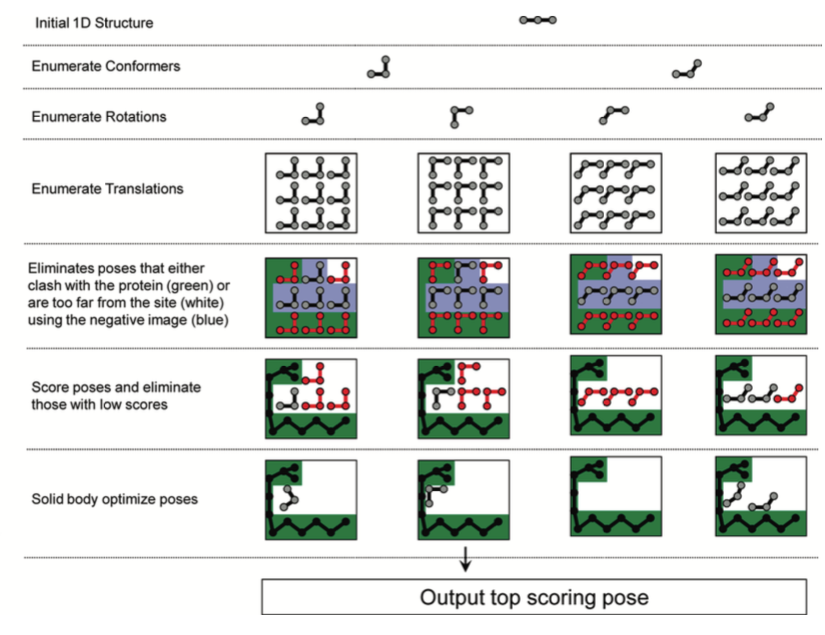
图1. FRED的算法,图片来源自文献3
在FRED对接的整个过程中,蛋白是刚性的,配体的每个构象也是刚性的,配体的柔性是隐式得包含在预先准备的配体构象系综中,即对接时输入的是预先构象搜索好的全部低能构象。因为化合物的构象生成不依赖于对接的结合位点,所以FRED分子对接的虚拟筛选极大地节省了对接计算所需的时间。在OEDocking的分子对接流程中,OMEGA用于对接前的构象搜索:根据Paul C. D. Hawkins等人的研究[5],OMEGA可以高效地搜索构象、并在低能构象中包含有一个生物活性构象(见图2)。
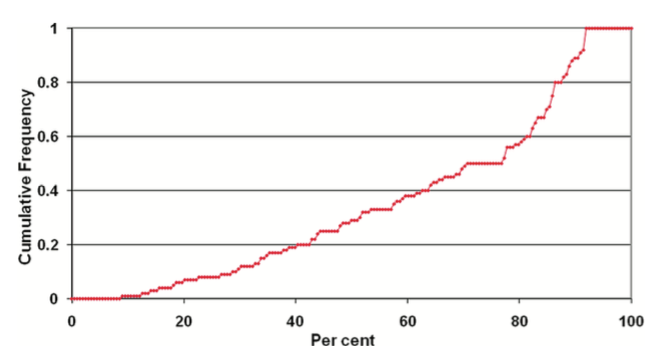
图2.在测试中,85%以上测试集的生物活性构象被OMEGA在1.5Å的误差内重现出来
FRED这种穷尽地分子对接方法非常高效。Brus等人[6]使用FRED发现了一种经过验证的BChE抑制剂(Ki = 2.7nM),作者将FRED描述为“迄今为止最快的对接工具,因此特别适用于超高通量对接。”
HYBRID是一种配体引导的分子对接,它用共晶配体的信息来提高虚拟筛选性能。与FRED一样,HYBRID对蛋白活性位点内的pose进行系统地、穷尽、非随机的检查。然而,HYBRID根据与已知共晶配体的形状和化学互补性来减少了搜索空间。与大多数对接程序相比,HYBRID这种配体引导的对接具有等效或更好的富集性能[3]。
HYBRID用靶标蛋白的结构及其活性位点内共晶的配体结构来预测配体的pose并进行打分。与FRED仅需apo蛋白结构不同的是,HYBRID在对接时需要用共晶配体的结构信息,由于蛋白通常在存在已知结合配体的情况下结晶,因此该信息通常可获得。如果同一种蛋白有不止一个共晶结构可用,HYBRID还能够用多个靶标蛋白构象进行对接。
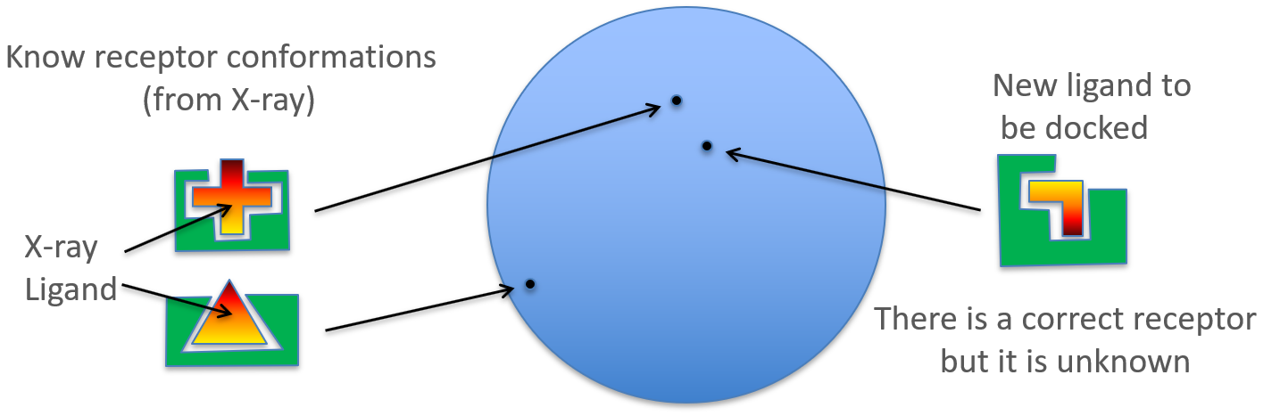
图3. 同一蛋白的同一结合位点与不同配体产生不同形状的结合口袋示意图。
FRED和HYBRID都使用穷尽的搜索算法来对接分子。在对接过程中,这两个程序都将配体构象作为刚性处理,通过对接每个配体的多个构象异构体来隐式地处理配体的柔性。在FRED和HYBRID的对接过程中,蛋白结构也被视为刚性;然而,HYBRID能够使用靶标蛋白的多个构象异构体,因此HYBRID也隐式地考虑了蛋白的柔性。
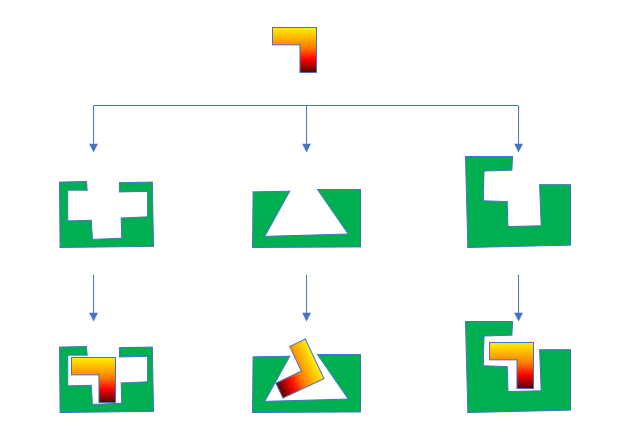
图4. 系综对接(Ensemble docking)示意图
如图3所示,当有多个蛋白-配体复合物结构可获取时,HYBRID实际上进行了系综对接(ensemble docking),首先将化合物与共晶配体进行形状与化学特征比较,然后将化合物对接到在形状与化学特征上最相似的共晶配体所在的蛋白结合位点里。这就有可能为一个新配体快速地从多个蛋白结合位点里找到最匹配的那个进行对接,而不必如图4所示的那样在逐个口袋进行对接计算。HYBRID比起FRED仅多花少量的计算量(15%),就可以完成系综对接获得更好的虚拟筛选性能[4]。
本文的目的是,以DUDE+[7]的PDE5A数据集为例,来演示使用单一蛋白结构的FRED分子对接虚拟筛选比之用了5个蛋白结构的HYBRID系综对接虚拟筛选性能上变化。更具体地讲,以ROC AUC、logAUC、BEDROC以及富集因子(Eenrichment Factor,EF)、富集比(Enrichment ratio 1%, ER1%)为指标,对DUDE+的PDE4A数据集上进行了性能评估,考察了两种方法在总体虚拟筛选性能与早期富集能力上的差异。还与AutoDock Vina 1.2,GOLD,GLIDE,Surflex-Dock、FlexX等软件在同一数据集上的性能表现进行了比较。需要强调的是,本文的目的不在于比较分子对接软件,而是想借助PDE5A这个算例来说明:信息用的更多的HYBRID方法,可以在不增加太多计算资源的情况下获得更好的性能。
2. 方法与材料
2.1 蛋白-配体共晶结构
从PDB下载1UDT、1UDU、3JWR、3SHZ以及3TGE等5个共晶结构,用OpenEye Applications 2021.02软件包中的spruce[8]进行蛋白结构准备。
Spruce是一款高精度蛋白结构准备软件,以便下游的应用软件使用。下游的应用包括:基团替换软件BROOD,分子对接软件FRED与HYBRID,结合模式预测软件POSIT以及结合位点水分子的位置与稳定性预测软件SZMAP等等。 Spruce结构准备的工作流程包括:1)将不对称单元扩展为生物单元;2)枚举可选的其它位置(alt locs);3)修复缺失的部分,比如侧链不完整、链断裂的封端以及缺失loop的建模;4)加氢原子及其优化,包括配体和辅酶的互变异构体枚举、以及对生物分子结构中互变异构体状态的评估;5)模型质量评估等等。
以PDB 1UDT共晶结构为例,说明如何用spruc进行e结构准备:
1 2 3 4 5 6 | $OE_DIR/bin/spruce -in 1udt.pdb -build_cterm_caps \ -build_loops \ -build_nterm_caps \ -build_sidechains \ -build_disulfidebridges \ -loop_db_filename /public/gkxiao/software/openeye/rcsb_spruce_1_0_20200422.loop_db |
spruce会自动识别设计单元,有的蛋白结构含有多个设计单元,则可能生成多个文件。
2.2 化合物数据库的准备
PDE5A的actives与decoys数据集(SDF格式)从DUDE[9]下载,用OpenEye Applications 2021.02软件包中的OMEGA[10]用默认参数进行构象搜索。
acitves化合物的结构准备:
1 2 3 4 5 6 7 | $OE_DIR/bin/oeomega pose -in actives_final.sdf \ -out actives.oeb.gz \ -flipper true \ -enumNitrogen true \ -enumRing true \ -prefix actives \ -useGPU |
decoys化合物的结构准备:
1 2 3 4 5 6 7 | $OE_DIR/bin/oeomega pose -in decoys_final.sdf \ -out decoys.oeb.gz \ -flipper true \ -enumNitrogen true \ -enumRing true \ -prefix decoys \ -useGPU |
构象搜索之后分别得到actives与decoys数据集的构象系综数据库:actives.oeb.gz与decoys.oeb.gz。
2.3 分子对接
OpenEye Applications 2021.02软件包中的FRED与HYBRID用于分子对接。其中FRED仅用1UDT准备的一个受体结构进行分子对接计算,而HYBRID以5个蛋白结构准备得到的受体结构进行分子对接计算。FRED与HYBRID分子对接虚拟筛选都采用默认参数进行计算,除了下面几个参数:
- mpi_np:24
- hitlist_size:0
- save_component_scores:true
- annotate_scores: true
其中mpi_np设置为24,意思是使用24个CPU核心进行并行计算。hitlist_size设为0是为了将所有化合物的计算结果都输出,而不是仅输出打分靠前指定数量的化合物。save_component_scores与annotate_scores设为true是希望输出的结果里,除了包含Chemgauss4打分之外,还将各种打分成分输出,以便在需要的时候可以考察打分成分对性能的影响,这本文中没有用到这个信息。
2.4 性能评估
鉴于DUDE下载的actives与decoys数据集因为互变异构体与质子化状态的枚举,导致一个化合物会以不同的形式多次出现、并可能被虚拟筛选多次命中,因此对虚拟筛选结果按化合物名称进行去重,同名化合物仅以其中打分最高那个来代表。所有的性能评估指标都在去重的基础上进行计算。此外,对接失败的化合物给予一个很高的打分值20。
AUC、logAUC、ER1%、三种α参数的BEDROC、富集因子(Enrichment Factor, EF)等指标用来评估虚拟筛选的性能,具体的计算方法见如何进行虚拟筛选的方法学验证[9]一文。
3. 结果
3.1 蛋白结构准备
Spruce对PDB 1udt、1udu、3jwr、3shz、3tge进行结构准备之后,得到如下几个准备好的蛋白结构:
1 2 3 4 5 6 7 8 | . ├── 1UDT_A__DU__VIA_A-1000.oedu ├── 1UDU_A__DU__CIA_A-1003.oedu ├── 1UDU_B__DU__CIA_B-2003.oedu ├── 3JWR_AC__DU__IBM_A-901.oedu ├── 3JWR_BD__DU__IBM_B-901.oedu ├── 3SHZ_A__DU__5CO_A-1.oedu └── 3TGE_A__DU__TGE_A-999.oedu |
其中1udu与3jwr因为包含有对称的A、B两个链,因此我们得到两个设计单元。
3.2 化合物数据库的准备
其中,actives数据包含398个不重复的化合物,经结构准备后为706个结构。全部结构都得到omega正确处理,状态信息如下:
1 2 3 4 5 6 | Number of Molecules Processed = 706 Number of Molecules Failed = 0 Average time/molecule = 0.226955 Average Number of Rotors = 6.395184 Average Number of Conformers = 316.879608 Elapsed Time = 160.229996 |
其中,decoys数据包含27521个不重复的化合物,经结构准备后为27969个结构,其中有20个没被处理,状态信息如下:
1 2 3 4 5 6 | Number of Molecules Processed = 27969 Number of Molecules Failed = 20 Average time/molecule = 0.201044 Average Number of Rotors = 7.034599 Average Number of Conformers = 356.125458 Elapsed Time = 5623.000000 |
因此,actives与decoy数据最终用于分子对接虚拟筛选的不重复的化合物数量分别为398与27501,虚拟筛选的性能根据不重复的化合物数来计算。而虚拟筛选的计算速度等其它场合,则用实际对接的各种形式的分子来处理,即acitves与decoys的化合物数分别按706与27949来计算。
3.3 FRED与HYBRID分子对接计算速度比较
FRED虚拟筛选仅利用一个apo蛋白结合位点信息,对actives化合物库进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 | $OE_DIR/bin/fred -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -dbase actives.oeb.gz\ -prefix actives_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
FRED对decoys化合物库进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 | $OE_DIR/bin/fred -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -dbase decoys.oeb.gz\ -prefix decoys_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
而HYBRID可以利用5个holo蛋白结合位点信息进行虚拟筛选,对actives进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 | $OE_DIR/bin/hybrid -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -receptor 1UDU_A__DU__CIA_A-1003.oedu\ -receptor 1UDU_B__DU__CIA_B-2003.oedu\ -receptor 3JWR_AC__DU__IBM_A-901.oedu\ -receptor 3JWR_BD__DU__IBM_B-901.oedu\ -receptor 3SHZ_A__DU__5CO_A-1.oedu\ -receptor 3TGE_A__DU__TGE_A-999.oedu\ -dbase actives.oeb.gz\ -prefix actives_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
同样的方式,用HYBRID对decoys进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 | $OE_DIR/bin/hybrid -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -receptor 1UDU_A__DU__CIA_A-1003.oedu\ -receptor 1UDU_B__DU__CIA_B-2003.oedu\ -receptor 3JWR_AC__DU__IBM_A-901.oedu\ -receptor 3JWR_BD__DU__IBM_B-901.oedu\ -receptor 3SHZ_A__DU__5CO_A-1.oedu\ -receptor 3TGE_A__DU__TGE_A-999.oedu\ -dbase decoys.oeb.gz\ -prefix decoys_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
FRED与HYBRID计算速度、分子处理情况可查阅对应的report文件或status文件,其中计算速度总结见表1。如表1所示,在同一台计算机上、用同样核心数,对同样的数据库进行虚拟筛选,虽然HYBRID在计算流程多了一步根据配体的信息进行蛋白选择,但是在这个例子里,HYBRID并没有比FRED计算速度更慢,相反还更快些。
表1、FRED与HYBRID在Actives与Decoys数据集上的计算速度比较
| 对接方法 | Actives每分子CPU时间 | Decoys每分子CPU时间 |
|---|---|---|
| FRED | 0.3 | 0.4 |
| HYBRID | 0.28 | 0.22 |
单位:秒
虽然在HYBRID筛选时提供了7个共晶结构,实际上HYBRID并没有对7个结合位点都进行对接。而是将待预测的化合物与7个共晶配体进行形状与化合物特征相似性比较,选择其中最相似的那个蛋白结合位点进行对接。因此,比之FRED,HYBRID仅是多花了基于配体的形状与药效团相似性比较步骤,但是这个计算非常快速。当HYBRID确定蛋白结合位点之后,尽管在对接计算算法上与FRED完全一致,然而HYBRID用CGO打分将搜索(分子的平动、转动)空间限制于比FRED更小空间内,这使得HYBRID需要搜索的空间更小,计算速度比FRED可能更快。
3.5 主要的虚拟筛选性能指标
用前文[9]描述的方法进一步计算AUC、logAUCλ=0.1%、ER1%、BEDROC、EF等评价虚拟筛选性能的参数,结果如表2所示。
表2、FRED单一受体结构分子对接与HYBRID多个受体结构的虚拟筛选性能比较
| 指标 | FRED | HYBRID |
|---|---|---|
| AUC | 0.79 | 0.82 |
| CI 95%(1000 resample) | 0.7597 – 0.8136 | 0.7970 – 0.8406 |
| ER1% | 27.7 | 32.2 |
| logAUCλ=0.1% | 43.5 | 47.1 |
| adjusted-logAUCλ=0.1% | 29.0 | 32.6 |
| BEDROC α=321.9 | 0.43 | 0.62 |
| BEDROC α=80.5 | 0.35 | 0.42 |
| BEDROC α=20.0 | 0.40 | 0.44 |
| EF0.5% | 30.0 | 43.1 |
| EF1% | 22.5 | 28.1 |
| EF5% | 8.1 | 8.6 |
| EF10% | 4.9 | 4.8 |
ROC AUC通常用来比较不同方法的总体虚拟筛选性能。如表2所示,HYBRID的ROC AUC略优于FRED的ROC AUC,两者的ROC曲线看起来没有明显的差异,如图5所示。
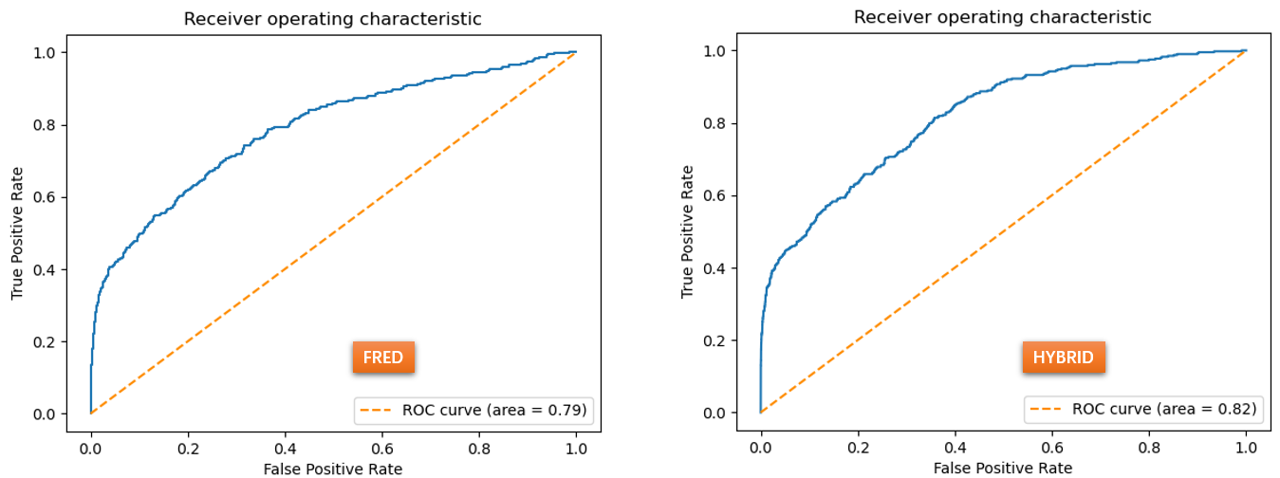
图 5. FRED与HYBRID分子对接的ROC曲线及其AUC
就PDE5A靶点而言,如图6所示,FRED的虚拟筛选总体性能(AUC=0.79)比Surflex(AUC=0.76)、DOCK(AUC=0.72)略优,而低于GLIDE(AUC=0.86);使用更多信息的HYBRID分子对接与Surflex系综对接,在AUC上略有提高,均为为0.82。
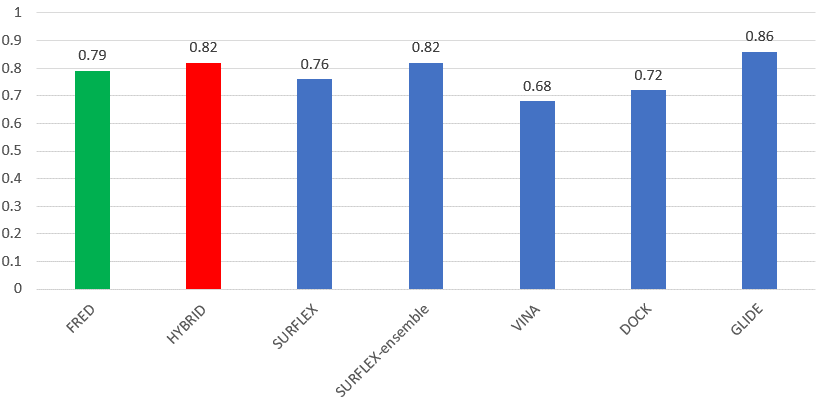
图 6. 不同分子对接虚拟筛选总体性能ROC AUC的比较
虽然使用了系综对接策略的HYBRID比单一蛋白对接的FRED在总体性能上提高幅度不大,但是在虚拟筛选早期富集能力上得到显著地提高,如表2所示,主要体现在HYBRID比FRED具有显著更高的logAUCλ=0.1%、ER1%、BEDROC、EF0.5%与EF1%。
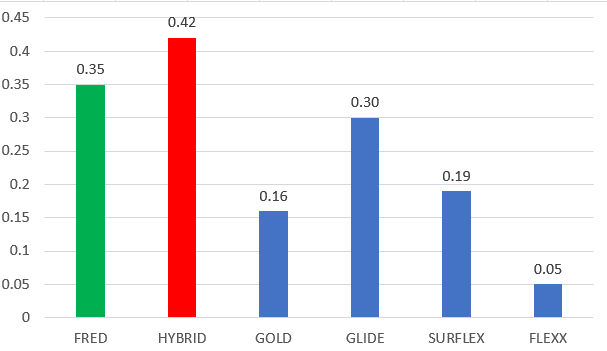
图 7. 不同分子对接虚拟筛选方法BEDROCα=80.5表征的早期富集能力比较
Chaput等人[12]用DUDE数据集对GOLD、GLIDE、Surflex与FLEXX进行了虚拟筛选性能比较。在他的研究中,用三种不同α参数的BEDROC来评估早期富集能力,其中当α=80.5时的性能数据可获取到。为了方便比较,我将Chaput等人[12]公开在PDE5A靶标上的性能指标与本文计算的BEDROCα=80.5性能指标一起汇总在图7。
如图7所示,使用了更多信息的HYBRID比FRED的早期富集性能大幅提高,BEDROCα=80.5分别为0.35与0.42。还发现,虽然GLIDE的总体性能比FRED与HYBRID好,但是就BEDROCα=80.5表征的早期富集能力而言,FRED与HYBRID却优于GLIDE,BEDROCα=80.5分别为0.35、0.42与0.30。在PDE5A这个靶标上,FRED与HYBRID的早期富集能力更是远优于GOLD、SURFLEX与FLEXX等流行的分子对接方法。
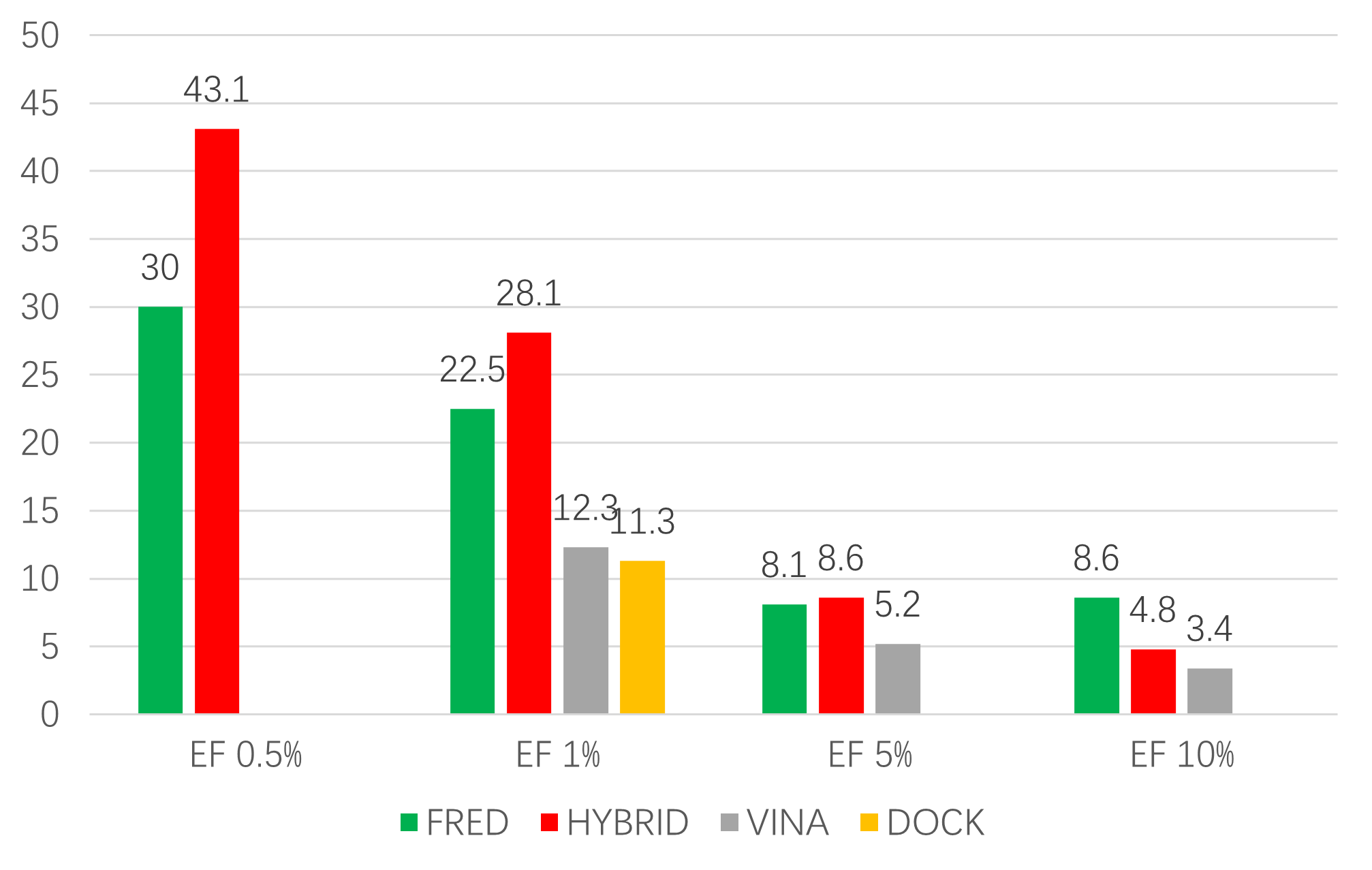
图 8. 不同分子对接虚拟筛选EF表征的早期富集能力比较
富集因子(EF)是另一常见的虚拟筛选性能评估方法。如图8所示,在以EF0.5%与EF1%表征的早期虚拟筛选筛选性能上,使用的更多信息的HYBRID分子对接要大幅领先于使用单一蛋白结合位点的FRED;而在以EF5%表征的不那么早期的虚拟筛选性能时,FRED与HYBRID相差不多;当以更不那么早期性能指标EF10%来评估时,FRED甚至优于HYBRID。
AutoDock Vina是最流行的免费、开源分子对接软件之一,其最新版本为Vina 1.2。Eberhardt等人[13]在DUDE上对AutoDock Vina 1.2进行了性能评估。为了方便比较,将Vina、FRED与HYBRID的性能指标汇总在图8。如图8所示,在PDE5A数据集上就EF1%、EF5%与EF10%表征的早期富集能力而言,FRED与HYBRID均大幅地优于Vina的12.25、5.16与3.43。总的来说,FRED与HYBRID比VINA不仅在虚拟筛选总体性能上优于Vina,而且在早期富集能力上优于Vina。
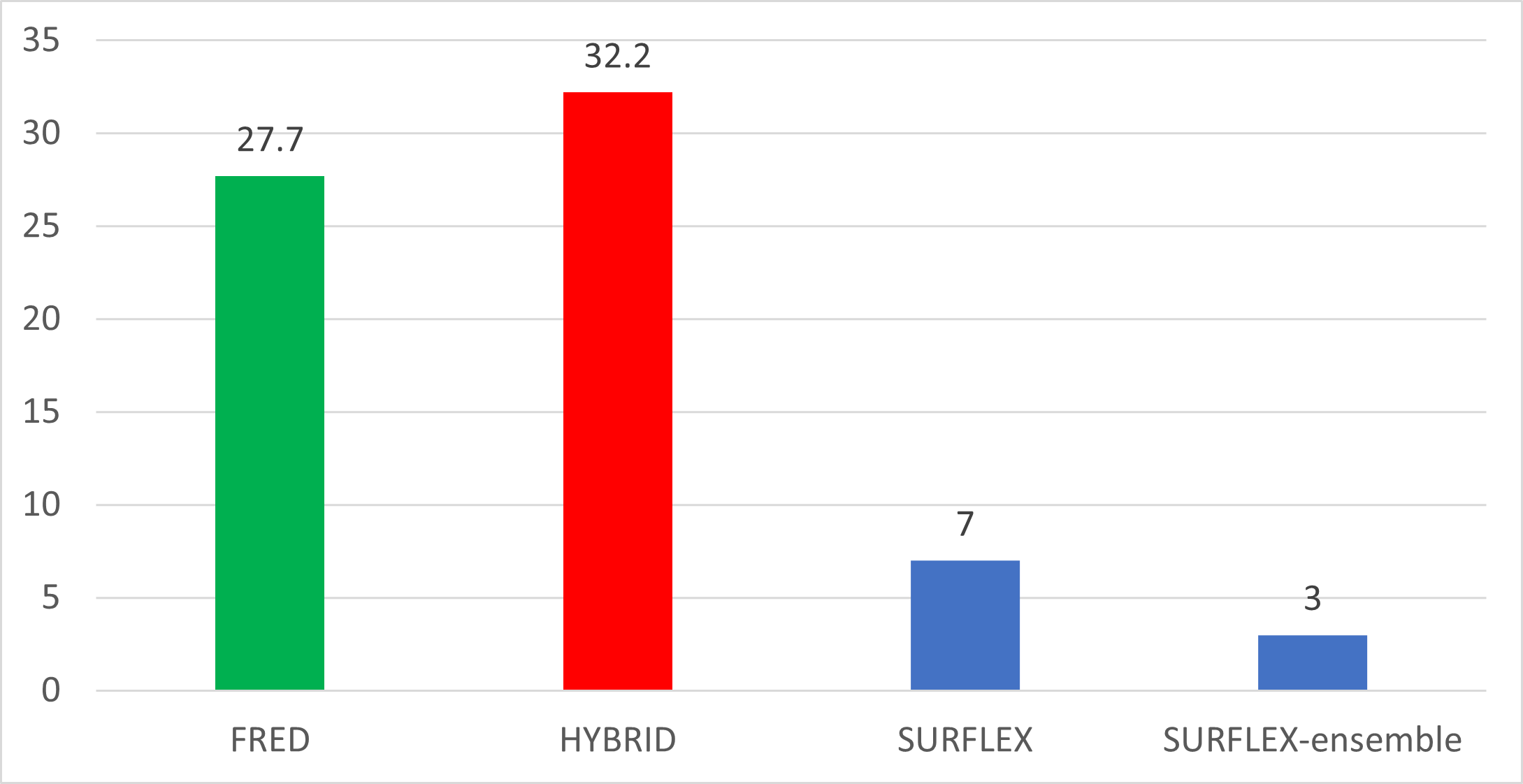
图 9. 不同分子对接虚拟筛选ER1%表征的早期富集能力比较
在Cleves等人的研究中[7],使用假阳性率为1%时的真阳性率百分比(ER1%)来评价早期富集能力,比较DUDE+92个靶标的数据,ER1%与BEDROCα=80.5有很好的相关性,虽然不是严格线性关系,但是为单调上升关系。HYBRID用到的受体与在Cleves等人研究中[7]的ensemble docking用的受体一致。因此,FRED/HYBRID与SURFLEX/SURFLEX-ensemble用ER1%来评估具有较好的可比性,结果如图9所示。
结果表明,虽然使用单一蛋白对接的FRED与SURFLEX虚拟筛选总体性能相当(AUC分别为0.79与0.76),但是FRED比SURFLEX具有显著更好的早期富集能力,体现在FRED比SURFLEX具有更大的ER1%,分别为27.7与7。相似地,使用多个蛋白信息的虚拟筛选HYBRID与Surflex-ensemble具有完全相同的虚拟筛选总体性能(AUC均为0.82),但是HYBRID比SURFLEX-ensemble具有显著更好的早期富集能力,体现在具有更大的ER1%,分别为32.2与3。当然,HYBRID的ER1%也比FRED的要高,再一次体现了:使用更多的信息,获得更好的性能。
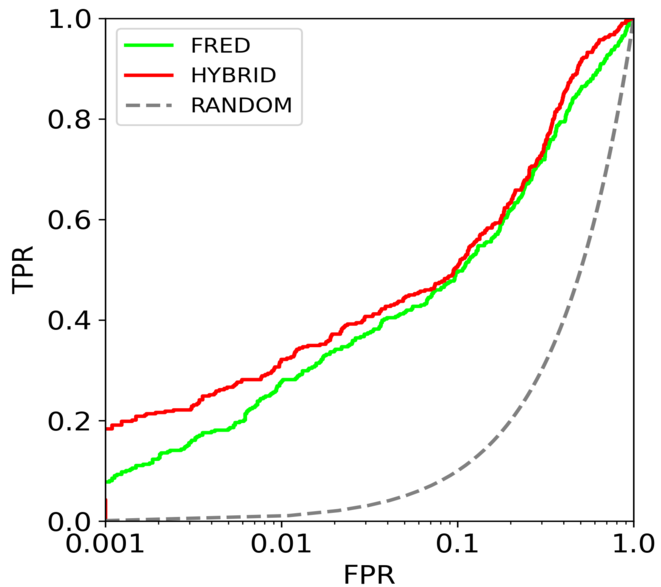
图 10. FRED与HYBRID的半对数ROC曲线,λ=0.001
前面提到,FRED与HYBRID的ROC曲线(见图5)看起来差不多,但是用表征早期富集能力的半对数ROC曲线(见图10)却可以发现显著的差异,这与计算的FRED与HYBRID虚拟筛选的logAUC值是一致的,logAUC分别为45.3%与47.1%(见表2)。
4.总结
FRED与HYBRID是OEDocking中两个基于分子对接的虚拟筛选工具。FRED和HYBRID都使用穷尽的搜索算法来对接分子。在对接过程中,这两个程序都将配体构象作为刚性处理,通过对接每个配体的多个构象异构体来隐式地处理配体的柔性。在FRED和HYBRID的对接过程中,蛋白结构也被视为刚性;然而,HYBRID能够使用靶标蛋白的多个构象异构体,因此HYBRID也隐式地考虑了蛋白的柔性。FRED仅使用一个apo蛋白结构进行分子对接,而HYBRID需要使用至少一个holo蛋白结构并与根据共晶配体进行形状与化学特征相似性为化合物选择一个合适的结合位点进行对接。
以ROC AUC为代表的整体性能而言,使用多个蛋白结构的HYBRID虚拟筛选比使用单一蛋白结构的FRED虚拟筛选的AUC略有提高,分别为0.79与0.82。HYBRID虚拟筛选在以logAUC、ER1%、BEDROCα=321.9、BEDROCα= 80.5、BEDROCα= 20.0、EF0.5%以及EF1%表征的早期富集性能上大幅优于FRED虚拟筛选。在不那么早期的EF5%与EF10%表征的虚拟筛选性能上,HYBRID与FRED相当。
在AUC表征的总体虚拟性能上,与流行的分子对接软件AutoDock Vina、DOCK、Glide与Surllex-Dock相比,在PDE5A这个数据集上,FRED虚拟筛选性能优于Surflex、VINA与DOCK,但比GLIDE差;而HYBRID表现出来的性能与同样利用了多个蛋白结构的Surflex-ensemble相当,优于使用单一蛋白对接的Surflex、Vina与DOCK,略低于GLIDE。这些不同的方法之间,AUC指标差异看起来不是特别的大。但是,在以BEDROCα=80.5为指标的虚拟筛选早期富集能力上,FRED与HYBRID却大幅领先于Gold、Glide、Surflex与FlexX;在以EF1%、EF5%与EF10%为指标的虚拟筛选早期富集能力上,FRED与HYBRID的性能也大幅高于AutoDock Vina。同样,在以ER1%为指标的虚拟筛选早期富集能力上,FRED与HYBRID虚拟筛选比Surflex-Dock单一蛋白或多个蛋白的系综虚拟筛选具有非常显著的性能优势。
最后,HYBRID虽然使用多个蛋白复合物结构进行虚拟筛选而FRED仅使用单一apo蛋白结构进行虚拟筛选,但是HYBRID并没有比FRED在计算速度有显著的损失。相反,在这个例子里,HYBRID对7个holo结构的虚拟筛选反而比单个apo结构进行对接的FRED计算速度更快。
总的来说,FRED利用一个apo蛋白进行分子对接虚拟筛选,而HYBRID支持使用多个holo蛋白结构进行对接虚拟筛选;HYBRID可以在不牺牲计算速度的情况下具有更好的虚拟筛选性能。因此,在有多个holo蛋白结构的情况下,强烈推荐使用HYBRID进行分子对接虚拟筛选。
5. 附件
数据集下载:oedocking_pde5a.tar.gz
下载后解压,共3个子目录,共46个文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 | . ├── database │ ├── actives.oeb.gz │ ├── actives.parm │ ├── actives.rpt │ ├── actives_status.txt │ ├── decoys.oeb.gz │ ├── decoys.parm │ ├── decoys.rpt │ └── decoys_status.txt ├── fred │ ├── 1UDT_A__DU__VIA_A-1000.oedu │ ├── 1udt.cif │ ├── 1udt.mtz │ ├── 1udt.pdb │ ├── actives_fred_docked.oeb.gz │ ├── actives_fred_report.txt │ ├── actives_fred_score.txt │ ├── actives_fred_settings.param │ ├── actives_fred_status.txt │ ├── crystal_ligand.mol2 │ ├── decoys_fred_docked.oeb.gz │ ├── decoys_fred_report.txt │ ├── decoys_fred_score.txt │ ├── decoys_fred_settings.param │ ├── decoys_fred_status.txt │ ├── decoys_fred_undocked.oeb.gz │ ├── ligand.mol2 │ ├── spruce_output.log │ └── spruce_settings.param └── hybrid ├── 1UDT_A__DU__VIA_A-1000.oedu ├── 1UDU_A__DU__CIA_A-1003.oedu ├── 1UDU_B__DU__CIA_B-2003.oedu ├── 3JWR_AC__DU__IBM_A-901.oedu ├── 3JWR_BD__DU__IBM_B-901.oedu ├── 3SHZ_A__DU__5CO_A-1.oedu ├── 3TGE_A__DU__TGE_A-999.oedu ├── actives_hybrid_docked.oeb.gz ├── actives_hybrid_report.txt ├── actives_hybrid_score.txt ├── actives_hybrid_settings.param ├── actives_hybrid_status.txt ├── decoys_hybrid_docked.oeb.gz ├── decoys_hybrid_report.txt ├── decoys_hybrid_score.txt ├── decoys_hybrid_settings.param ├── decoys_hybrid_status.txt ├── decoys_hybrid_undocked.oeb.gz └── receptor.lst 3 directories, 46 files |
其中database目录的actives.oeb.gz与decoys.oeb.gz,是OMEGA从DUDE的actives与decoys的sdf开始准备的化合物库;其中fred与hybrid目录分别包含了两种分子对接计算所需的受体文件与对接结果。
6. 文献
- OEDocking. https://www.eyesopen.com/oedocking
- McGann, M. R.; Almond, H. R.; Nicholls, A.; Grant, J. A.; Brown, F. K. Gaussian Docking Functions. Biopolymers 2003, 68 (1), 76–90. https://doi.org/10.1002/bip.10207.
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51 (3), 578–596. https://doi.org/10.1021/ci100436p.
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided. Mol. Des. 2012, 26 (8), 897–906. https://doi.org/10.1007/s10822-012-9584-8.
- Hawkins, P. C. D.; Skillman, A. G.; Warren, G. L.; Ellingson, B. A.; Stahl, M. T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50 (4), 572–584. https://doi.org/10.1021/ci100031x.
- Brus, B.; Košak, U.; Turk, S.; Pišlar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J. P.; Gobec, S. Discovery, Biological Evaluation, and Crystal Structure of a Novel Nanomolar Selective Butyrylcholinesterase Inhibitor. J. Med. Chem. 2014, 57 (19), 8167–8179. https://doi.org/10.1021/jm501195e.
- Cleves, A. E.; Jain, A. N. Structure- and Ligand-Based Virtual Screening on DUD-E + : Performance Dependence on Approximations to the Binding Pocket. J. Chem. Inf. Model. 2020, 60 (9), 4296–4310. https://doi.org/10.1021/acs.jcim.0c00115.
- Spruce. https://www.eyesopen.com/spruce
- DUDE的PDE5a数据集. 2022-01-25访问. http://dude.docking.org/targets/pde5a
- OMEGA. https://www.eyesopen.com/omega
- 肖高铿. 如何进行虚拟筛选的方法学验证. 墨灵格的博客. 2016-09-22. http://blog.molcalx.com.cn/2016/09/22/virtual-screening-methodology-validation.html
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of Four Popular Virtual Screening Programs: Construction of the Active/Decoy Dataset Remains a Major Determinant of Measured Performance. J. Cheminform. 2016, 8 (1), 56. https://doi.org/10.1186/s13321-016-0167-x.
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, acs.jcim.1c00203. https://doi.org/10.1021/acs.jcim.1c00203.


