
OEDocking——信息用的越多,虚拟筛选性能越好
摘要:本文用DUDE+数据集对OEDocking使用单一apo结构的FRED分子对接、单一holo结构的HYBRID分子对接(HYBRID-S)以及多个holo结构的HYBRID分子对接(HYBRID-M)虚拟筛选性能进行了评估。结果表明,在以AUC为代表的总体虚拟筛选性能上,三者没有显著的差异;在以BEDROC、EF为代表的早期富集能力上,虚拟筛选性能从高到低依次为:HYBRID-M、HBYRID-S、FRED。这与信息用的越多性能越好的预期一致。还与几个流行的分子对接软件GLIDE与GOLD等进行了比较,结果发现,在EF0.5%、EF1%、或BEDROCα=321.9等表征的早期识别能力指标上,虚拟筛选性能从高到低排序依次为:HYBRID-M、GLIDE与HYBIRD-S相当、FRED、GOLD。
肖高铿/2022-03-21
1. 简介
FRED与HYBRID是OpenEye OEDocking软件包中两款分子对接虚拟筛选工具[1]。在FRED对接的整个过程中,蛋白是刚性的,配体的每个构象也是刚性的,配体的柔性是隐式得包含在预先准备的配体构象系综中,即对接时输入的是预先构象搜索好的全部低能构象。HYBRID是一种配体引导的分子对接,它用共晶配体的信息来提高虚拟筛选性能。与FRED一样,HYBRID对蛋白活性位点内的pose进行系统地、穷尽、非随机的检查。然而,HYBRID根据与已知共晶配体的形状和化学互补性来减少了搜索空间。与大多数对接程序相比,HYBRID这种配体引导的对接具有等效或更好的富集性能[2-4]。如果同一种蛋白有不止一个共晶结构可用,HYBRID还能够用多个靶标蛋白构象进行对接。
FRED和HYBRID都使用穷尽的搜索算法来对接分子。在对接过程中,这两个程序都将配体构象作为刚性处理,通过对接每个配体的多个构象异构体来隐式地处理配体的柔性。在FRED和HYBRID的对接过程中,蛋白结构也被视为刚性;然而,HYBRID能够使用靶标蛋白的多个构象异构体,因此HYBRID也隐式地考虑了蛋白的柔性。
McGann等人[4]在多个受体版本的DUD(Multi receptor DUD, MDUD)数据集上证明了:使用了配体信息的HYBRID比仅使用蛋白apo结构FRED具有更好的虚拟筛选性能;HYBRID使用多个蛋白-配体复合物结构比仅使用一个蛋白-配体复合物结构的具有更好的虚拟筛选性能。也就是说:信息用的越多,虚拟筛选性能越好。
最近,UCSF的Jain课题组[5]开发了DUDE+数据集用来评估使用多个蛋白结构与使用单一蛋白结构基于分子对接/基于配体虚拟筛选方法的性能;除了ROC AUC之外,而且还引入了ER 1%指标来评估虚拟筛选方法的早期富集能力。本的主要目的之一是用DUDE+来评估:FRED使用单一蛋白apo结构、HYBRID使用单一holo结构以及HYBRID使用多个holo结构进行虚拟筛选的性能差异。
2. 方法与材料
2.1 DUDE+数据集的蛋白-配体共晶结构准备
DUDE+是DUDE的一个子集,包含了92个靶标。每个靶标除了包含了一个与DUDE一样的蛋白-配体复合物结构之外,还包含了同一个靶标之外的5个蛋白配体复合物结构(部分复合物结构有重复)。典型的相关文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | [gkxiao@master pde5a]$ tree . ├── alt-lig1.mol2 ├── alt-lig2.mol2 ├── alt-lig3.mol2 ├── alt-lig4.mol2 ├── alt-lig5.mol2 ├── alt-pro1.mol2 ├── alt-pro2.mol2 ├── alt-pro3.mol2 ├── alt-pro4.mol2 ├── alt-pro5.mol2 ├── ligand.mol2 ├── protein.mol2 |
其中ligand.mol2与protein.mol2是与DUDE一致的蛋白-配体复合物结构准备而来。除此之外,还包含alt-lig与alt-pro开头的5个配体、受体文件,分别来自5个蛋白配体复合物结构。有时,alt中一个会与DUDE中的那个复合物结构重复。以PDE5A为例,ligand.mol2与protein.mol2来自PDB 1UDT;其它的5个alt分别来自5个PDB结构:1UDT,1UDU、3JWR、3SHZ以及3TGE。其中,1UDT就重复出现了。
每个复合物结构进一步用OpenEye Applications 2021.02软件包中的spruce[6]进行蛋白结构准备。Spruce是一款高精度蛋白结构准备软件,以便下游的应用软件使用。Spruce结构准备的工作流程包括:1)将不对称单元扩展为生物单元;2)枚举可选的其它位置(alt locs);3)修复缺失的部分,比如侧链不完整、链断裂的封端以及缺失loop的建模;4)加氢原子及其优化,包括配体和辅酶的互变异构体枚举、以及对生物分子结构中互变异构体状态的评估;5)模型质量评估等等。
以PDB 1UDT共晶结构为例,说明如何用spruc进行蛋白结构准备:
1 2 3 4 5 6 | $OE_DIR/bin/spruce -in 1udt.pdb -build_cterm_caps \ -build_loops \ -build_nterm_caps \ -build_sidechains \ -build_disulfidebridges \ -loop_db_filename /public/gkxiao/software/openeye/rcsb_spruce_1_0_20200422.loop_db |
spruce会自动识别设计单元,有的蛋白结构含有多个设计单元,则可能生成多个文件。在本测试中,每个PDB结构仅选择其中一个作为对接用的蛋白结构。以PDE5A为例,Spruce对PDB 1udt、1udu、3jwr、3shz、3tge进行结构准备之后,得到如下5个准备好的蛋白结构(设计单元):
1 2 3 4 5 6 | . ├── 1UDT_A__DU__VIA_A-1000.oedu ├── 1UDU_A__DU__CIA_A-1003.oedu ├── 3JWR_AC__DU__IBM_A-901.oedu ├── 3SHZ_A__DU__5CO_A-1.oedu └── 3TGE_A__DU__TGE_A-999.oedu |
这些.oedu的设计单元文件可直接给FRED或HYBRID进行虚拟筛选。
2.2 DUDE+化合物数据库的准备
DUDE+的active、decoy化合物与DUDE[7]中的完全一样。以其中PDE5A靶标下的actives与decoys数据集为例来说明如何进行结构准备。首先从从DUDE下载SDF格式文件,用OpenEye Applications 2021.02软件包中的OMEGA[8]用默认参数进行构象搜索。
acitves化合物的结构准备:
1 2 3 4 5 6 7 | $OE_DIR/bin/oeomega pose -in actives_final.sdf \ -out actives.oeb.gz \ -flipper true \ -enumNitrogen true \ -enumRing true \ -prefix actives \ -useGPU |
decoys化合物的结构准备:
1 2 3 4 5 6 7 | $OE_DIR/bin/oeomega pose -in decoys_final.sdf \ -out decoys.oeb.gz \ -flipper true \ -enumNitrogen true \ -enumRing true \ -prefix decoys \ -useGPU |
构象搜索之后分别得到actives与decoys数据集的构象系综数据库:actives.oeb.gz与decoys.oeb.gz。
2.3 分子对接
OpenEye Applications 2021.02软件包中的FRED与HYBRID用于分子对接。FRED与HYBRID分子对接虚拟筛选都采用了相同的默认参数进行计算,除了下面几个参数:
- mpi_np:24
- hitlist_size:0
- save_component_scores:true
- annotate_scores: true
其中mpi_np设置为24,意思是使用24个CPU核心进行并行计算。hitlist_size设为0是为了将所有化合物的计算结果都输出,而不是仅输出打分靠前指定数量的化合物。save_component_scores与annotate_scores设为true是希望输出的结果里,除了包含Chemgauss4打分之外,还将各种打分成分输出,以便在需要的时候可以考察打分成分对性能的影响,这本文中没有用到这个信息。
此外,为了考察FRED、HYBRID的性能与信息利用程度之间的关系,设计三组对接计算,以PDE5A为例示范如下:
2.3.1 FRED分子对接虚拟筛选,仅用DUDE蛋白-配体复合物的受体结构进行虚拟筛选
FRED仅利用一个apo蛋白结合位点信息对actives化合物库进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 | $OE_DIR/bin/fred -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -dbase actives.oeb.gz\ -prefix actives_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
FRED对decoys化合物库进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 | $OE_DIR/bin/fred -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -dbase decoys.oeb.gz\ -prefix decoys_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
2.3.2 HYBRID仅用一个holo结构进行虚拟筛选,标记为HYBRID-S
HYBRID仅利用DUDE的一个holo结构对actives化合物库进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 | $OE_DIR/bin/hybrid -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -dbase actives.oeb.gz\ -prefix actives_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
HYBRID仅利用DUDE的一个holo结构对decoys化合物库进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 | $OE_DIR/bin/hybrid -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -dbase decoys.oeb.gz\ -prefix decoys_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
2.3.3 HYBRID用DUDE+中多个holo结构进行虚拟筛选,标记为HYBRID-M
HYBRID利用多个holo蛋白结合位点信息对actives进行虚拟筛选,在PDE5A靶标里有5个holog结构,命令如下:
1 2 3 4 5 6 7 8 9 10 11 | $OE_DIR/bin/hybrid -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -receptor 1UDU_A__DU__CIA_A-1003.oedu\ -receptor 3JWR_AC__DU__IBM_A-901.oedu\ -receptor 3SHZ_A__DU__5CO_A-1.oedu\ -receptor 3TGE_A__DU__TGE_A-999.oedu\ -dbase actives.oeb.gz\ -prefix actives_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
同样的方式,用HYBRID对decoys进行虚拟筛选,命令如下:
1 2 3 4 5 6 7 8 9 10 11 | $OE_DIR/bin/hybrid -mpi_np 24\ -receptor 1UDT_A__DU__VIA_A-1000.oedu\ -receptor 1UDU_A__DU__CIA_A-1003.oedu\ -receptor 3JWR_AC__DU__IBM_A-901.oedu\ -receptor 3SHZ_A__DU__5CO_A-1.oedu\ -receptor 3TGE_A__DU__TGE_A-999.oedu\ -dbase decoys.oeb.gz\ -prefix decoys_fred\ -hitlist_size 0\ -save_component_scores true\ -annotate_scores true |
2.4 性能评估
鉴于DUDE下载的actives与decoys数据集因为互变异构体与质子化状态的枚举,导致一个化合物会以不同的形式多次出现、并可能被虚拟筛选多次命中,因此对虚拟筛选结果按化合物名称进行去重,同名化合物仅以其中打分最高那个来代表。所有的性能评估指标都在去重的基础上进行计算。此外,对接失败的化合物给予一个很高的假想打分值(20)以确保active与decoy的数量与DUDE中的一致,这也是性能评估参数具有可比性的前提。
AUC、logAUC(指调整过的logAUC)、三种α参数的BEDROC、富集因子(Enrichment Factor, EF)等指标用来评估虚拟筛选的性能,具体的计算方法见如何进行虚拟筛选的方法学验证[9]一文。
3. 结果
3.1 在DUDE+数据集上的AUC与logAUC比较
如图1所示,在DUDE+数据集上,FRED对接虚拟筛选、使用单一受体的HYBRID对接虚拟筛选(HYBIRD-S)与使用多个受体的HYBRID对接虚拟筛选(HYBRID-M)在92个靶标上AUC的平均值/中值分别为0.79/0.79、0.78/79与0.81/81,Friedman Test检验statistic=4.717且p=0.094,因此认为FRED、HYBRID-S与HYRID-M的AUC均值间没有统计学意义上的显著性差异。
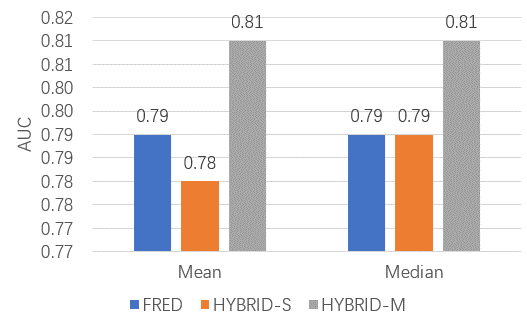
图1. FRED、HYBRID-S(一个受体结构)与HYBRID-M(多个受体结构)虚拟筛选的AUC均值与中值
这个结论与McGann等人[4]在MDUD(Multi-receptor DUD,MDUD)数据集上测试得到的AUC性能表现不同。在McGann等人[4]的研究中,FRED、HYBRID-S与HYBRID-M三种方法的AUC均值具有显著的差异,虚拟筛选性能从高到低排序依次为:HYBRID-M、HYBRID-S与FRED。因此,在不同的数据集上,可能得到不同的性能比较结论。
表1. 多种分子对接虚拟筛选的AUC值分布
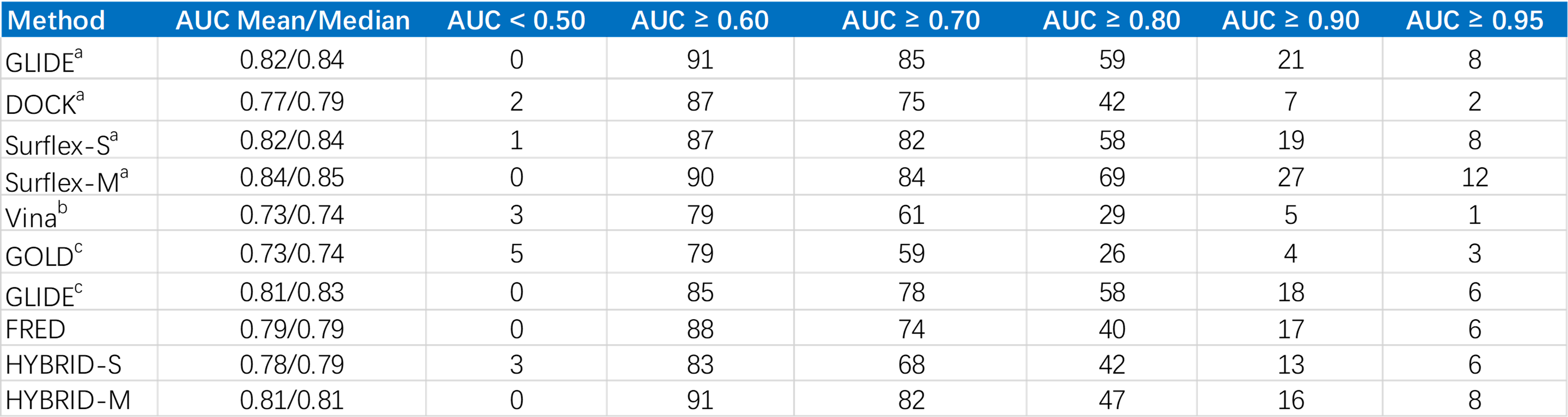
a:文献5,Surflex-S表示单一受体对接,Surflex-M表示多个受体对接; b: 文献11; c: 文献10(按2.4小节的方法统计而来)
表1汇集了多种分子对接虚拟筛选方法在DUDE+数据集上的AUC均值、中值以及在不同区间的分布。除了VINA与GOLD明显的差于其它方法之外,FRED与HYBRID似乎没有特别之处。但是,如表2所示,用半对数ROC曲线下面积(logAUCλ=0.001——一种早期富集能力得到强调的评价指标——似乎可以看到不同的趋势。
表2. 多种对接虚拟筛选的logAUCλ=0.001值分布

首先观察FRED、HYBRID-S与HYRID-M的logAUCλ=0.001均值/中值,分别为0.315/0.263、0.323/0.290、0.350/0.328,如图2所示,三者之间貌似有数值差异,并且呈现从低到高的趋势。但经过Friedman Test检验,statistic=6.021且p=0.05,因此认为FRED、HYBRID-S与HYRID-M的logAUC均值间没有统计学意义上的显著性差异。这也提示我们需要用EF与BEDROD等专门评估早期富集能力的指标来进一步评价这些方法。
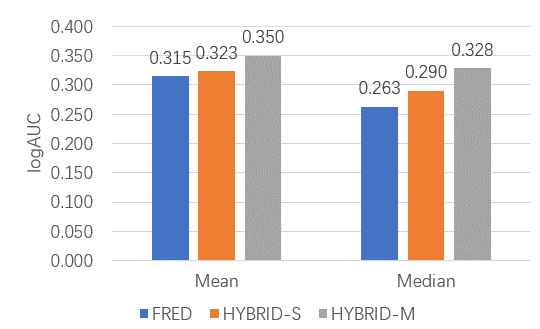
图2. FRED、HYBRID-S(一个受体结构)与HYBRID-M(多个受体结构)虚拟筛选的logAUCλ=0.001均值与中值
如5.1小节的统计学分析所示,Friedman假设检验提示在FRED、HYBRID-S、HYBRID-M、DOCK、GOLD与GLIDE的logAUC之间至少有一个方法与其它方法存在统计学意义上的显著差异(statistic=94.851, p-value=6.418e-19)。进一步用Nemenyi post-hoc test进行分析,可以发现GOLD的虚拟筛选性能显著地差于其它五种分子对接虚拟筛选方法,HYBRID-M显著地优于DOCK方法。
3.2 在DUDE+数据集上四种不同通量水平(X=0.5%、1%、5%、10%)时的富集因子EFX比较
图3呈现了在不同通量水平下(打分最高0.5%、1%、5%、10%化合物)不同分子对接方法的富集因子累积分布图,越上方的曲线具有更多的算例数优于特定的EF截断值,也就是具有更好的富集能力。蓝色水平线为EF=1,所有方法在大部分算例上的EF都大于1,因此所有方法都具有一定的活性化合物富集能力。
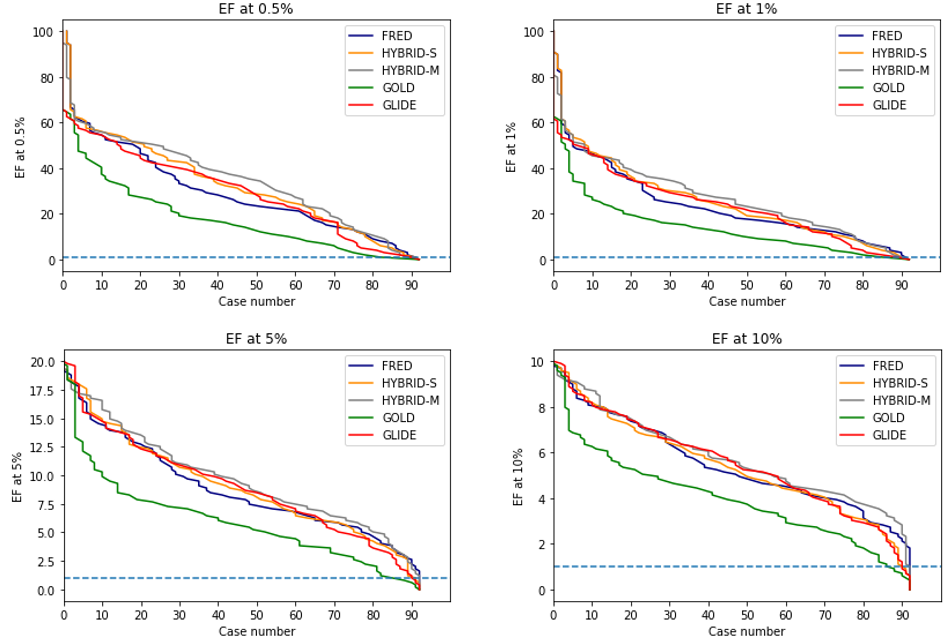
图3. FRED、HYBRID-S、HYBRID-M、GOLD与GLIDE虚拟筛选方法在4种不同通量水平的EF值分布比较,其中蓝色水平线为EF=1
根据图3,可以发现,无论是在哪个通量水平下,GOLD(绿色)曲线总在其它曲线下方,这说明GOLD的富集能力比其它四中方法的差,因此性能排名最末,这一点与根据AUC值得出的结论一致。
此外,灰色的HYBRID-M曲线基本总在其它分子对接方法的上方,这说明使用了多个受体结构进行对接的HYBRID-M具有最佳的富集能力。但是也发现,HYBRID-M的优势随着通量水平的提高而降低。在较低通量时(top 0.5%与1%),HYBRID-M与其它方法曲线间的间隔大;随着通量的提高(top 5%与10%),与其它方法曲线间的间隔变小。比如,在top 10%的通量水平,FRED、GLIDE,HYBRID-S、HYBRID-M四条曲线几乎完全纠缠在一起,难以区分。
无论在哪个通量水平,HYBRID-S与GLIDE曲线纠缠最厉害,因此可以这两种方法的富集能力差不多;而相比之下深蓝色的FRED曲线略微位于HYBRID-S与GLIDE的下方,因此可以认为FRED的性能低于前面两者。
综上所述,从不同通量水平的富集因子来说,所有方法对大部分靶标EF都大于1,因此都表现出了活性化合物的富集能力。不同方法的富集性能从高到低排序依次如下:HYBRID-M,GLIDE与HYBRID-S相似,FRED,GOLD。富集性能与通量水平有关系,当考虑top 0.5%的通量水平时,HYBRID-M显著优于其它,随着通量的提高,HYBRID-M的富集能力优势缩小。就FRED、HYBRID-S与HYBRID-M而言,EF表征的性能依次提高,体现了信息用的越多、虚拟筛选性能越好的规律。
3.3 在DUDE+数据集上的BEDROC比较
不同α参数的BEDROC代表了不同通量水平的富集能力,根据Chaput等人[13]的研究表明,α=321.9、80.5、20.0时BEDROC分别与打分靠前0.5%、2%、8%的EF所表示的性能相当。图4绘制了三种α参数下BEDROC与算例数的曲线图,其中蓝色水平线代表BEDROC=0.5。
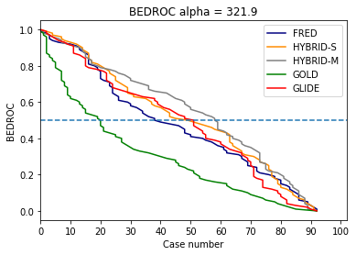

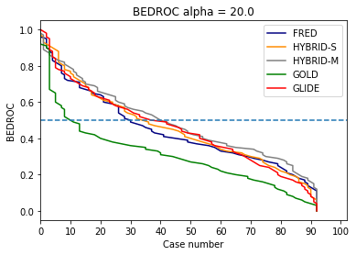
图4. 五种分子对接方法BEDROC(三种α值)比较,不低于0.5的靶标数越多的方法性能越好,蓝色水平线为BEDROC=0.5。
从图4可以看到,当α=321.9时,HYBRID-M明显地位于其它曲线的上方,而GOLD明显地位于其它曲线的下方,HYBRID-S与GLIDE纠缠在一起难分上下,而FRED则位于HYBRID-S与GLIDE之下。因此,在α=321.9时,BEDROC表征的虚拟筛选性能排序从高到低为:HYBRID-M、GLIDE与HYBRID-S相当、FRED、GOLD。
当α=80.5与20的时候,不同方法的性能顺序大体与之前α=321.9时一致,但是HYBRID-M曲线与HYBRID-S、GLIDE的曲线间的间隙变小,这说明HYBRID-M的性能优势随着α的减小而变小。这种变化趋势与EF随通量水平变化是一致的,也与Chaput等人[13]的研究结果一致。
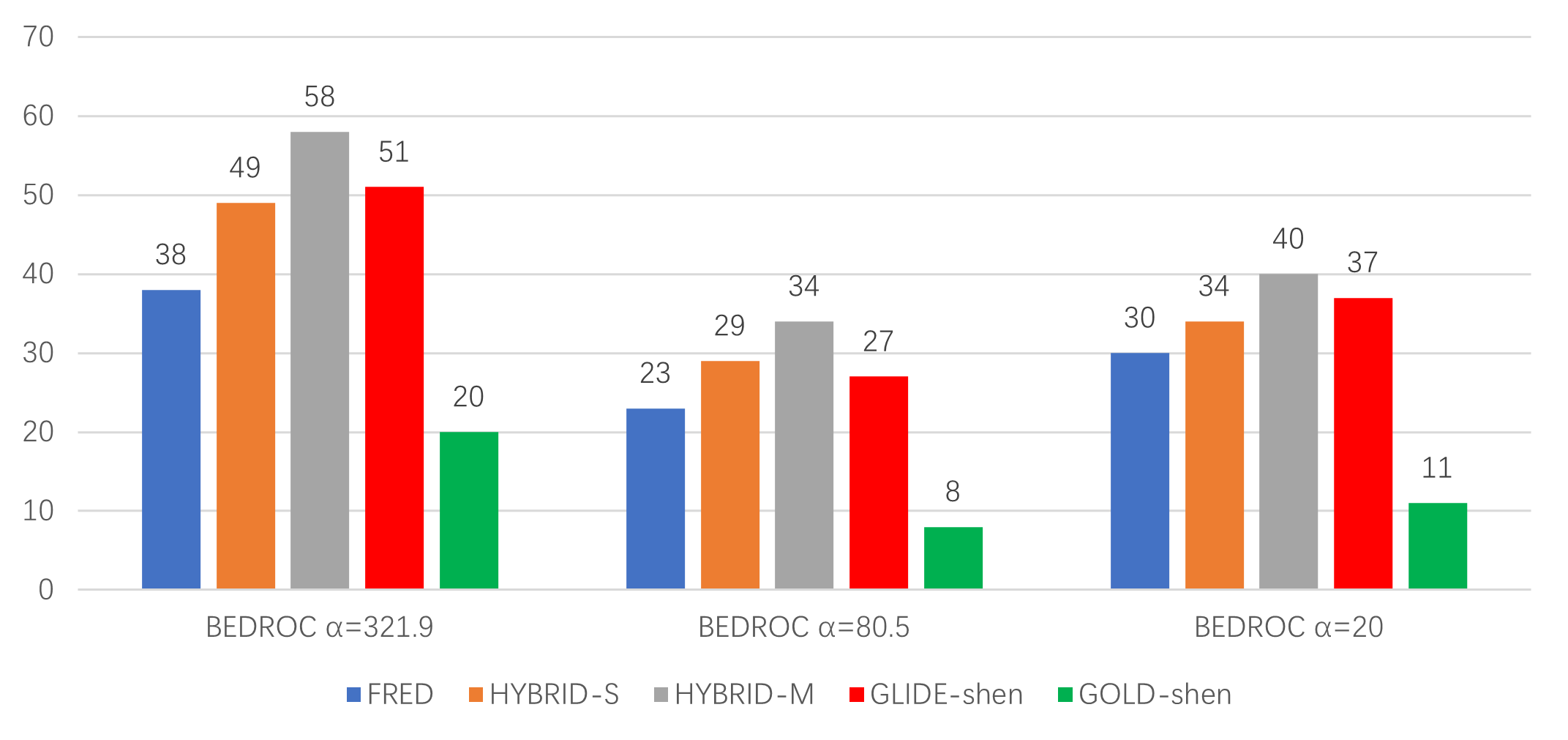
图5. 五种分子对接方法BEDROC(三种α值)不低于0.5的靶标数比较。靶标数越大的方法,性能越好。
将BEDROC不小于0.5视为虚拟成功,统计不同方法在三种α值时成功虚拟筛选的算例数,结果如图5所示。当α=321.9时,不同方法成功虚拟筛选的算例数从高到低排序依次为:HYBRID-M(58)、GLIDE(51)、HYBRID-S(49)、FRED(38)、GOLD(20);当α=80.5时,不同方法成功虚拟筛选的算例数从高到低排序依次为:HYBRID-M(34)、HYBRID-S(29)、GLIDE(27)、FRED(23)、GOLD(8);当α=20时,不同方法成功虚拟筛选的算例数从高到低排序依次为:HYBRID-M(40)、GLIDE(37)、HYBRID-S(34)、FRED(30)、GOLD(11)。可见虚拟筛选性能排序随着α变化而变化。但总的来说,HBYRID-M性能总是最佳;之后为GLIDE与HYBRID,相差不多;在接着是FRED;GOLD最后。就FRED、HYBRID-S与HYBRID-M而言,以BEDROC不小于0.5的靶标数表示的性能依次从低到高,再一次体现了信息用的越多、虚拟筛选性能越好的规律。

图6. 当α=321.9时,HYBRID-M与GLIDE BEDROC不低于0.5的靶标数比较
需要注意的是,虚拟筛选成功的靶标数评价的是整体性能,并不意味着一个方法比另一个方法在各个靶标上都具有优势。如图6所示,当α=321.9时,HYBRID-M在58个算例上虚拟筛选成功,而GLIDE在51个算例上虚拟筛选成功。这两种方法都在同样的40个算例上虚拟筛选成功,在HYBRID-M虚拟筛选成功中,有18个算例是GLIDE虚拟筛选失败的;而在GLIDE虚拟筛选成功中,有11个算例是HYBRID-M失败的。因此HYBRID-M与GLIDE两者的关系是互补的,联合起来可在69个算例上虚拟筛选获得成功,高于任何一种单一的方法。实际上,通过数据融合策略联用不同方法提高虚拟筛选性能已被证明是有效的[14]。

图7. 当α=321.9时,HYBRID-M与FRED BEDROC不低于0.5的靶标数比较
相比之下,FRED与HYBRID的虚拟筛选算法基本一样,只是利用的信息不同,两者间的性能互补性不像HYBRID与GLIDE间那么强。如图7所示,当α=321.9时,HYBRID-M在58个算例获得成功,而FRED在38个算例上虚拟筛选成功。这两种方法都在同样的36个算例上虚拟筛选成功,除了两个靶标之外,HYBRID-M虚拟筛选基本完全覆盖了FRED虚拟筛选成功的靶标。这说明HYBRID与FRED之间基本没有互补性,而只是体现了:信息用的越多、虚拟筛选性能越好。
3.4 计算速度
HYBRID与FRED都是用预先搜索好构象的配体进行刚性对接,配体的柔性隐含在预先搜索好的配体构象里,因此HYBRID与FRED对接计算的效率非常高。之前在DUDE PDE5A数据集上已经讨论过[15],请读者自行阅读,本文不再叙述。
4. 结论
在本文中,我们在DUDE+数据集上测试了OEDocking中的FRED与HYBRID的虚拟筛选性能,其中将仅用一个受体的HYBRID虚拟筛选标记为HYBRID-S,使用了多个受体进行虚拟筛选的HYBRID标记为HYBRID-M。FRED、HYBRID-S、HYBRID-M的分子对接对接算法完全相同,区别在于FRED仅利用受体结构信息(apo结构);虽然HYBRID与FRED使用同样的受体结构信息,但是还用了结合位点里的配体信息进行形状比较,也就是用了holo结构信息;HYBRID-M则同时使用了几个holo结构。总的来说,FRED,HYBRID-S,HYBRID-M依次使用了更多的信息进行于分子对接虚拟筛选。虚拟筛选性能用AUC、logAUC,富集因子EF与BEDROC来进行评价。
结果表明,以AUC表征的虚拟筛选性能,不能区分FRED,HYBRID-S与HYBRID-M三者之间的性能差异;用放大了早期富集能力的半对数AUC(logAUC)来进行评价,从均值上看,性能从高到低依次为HYBRID-M、HYBRID-S、FRED,但Friedman Test假设检验分析表明,并不具备统计学意义上的显著差异。
在不同通量水平(top 0.5%, 1%, 5%与10%)计算的富集因子(EF)表明,当筛选通量比较低时,EF0.5%与EF1%表征富集能力从高到低排序依次为:HYBRID-M、HYBRID-S与GLIDE相当、FRED、GOLD;随着通量的增加(top 5%,10%),HYBRID-M相对HYBRID-S与GLIDE的优势降低。就FRED、HYBRID-S与HYBRID-M而言,不论通量水平,性能总是依次从低到高,体现了信息用的越多、筛选性能越好。
三种不同α参数(321.9、80.5、20.0)BEDROC表征的富集能力分析表明,当α=321.9时(相当于top 0.5%通量水平),虚拟筛选性能从高到低依次为:HYBRID-M、GLIDE与HYBRID-S相当、FRED、GOLD;随着通量的加大,BEDROC表征的性能差异也缩小。就FRED、HYBRID-S与HYBRID-M而言,不论α参数的值,性能总是依次从低到高,体现了信息用的越多、筛选性能越好。
BEDROC分析还表明,不同的分子对接方法之间具有显著的虚拟筛选性能互补性,比如HYBRID-M与GLIDE就具有互补性,联用两种方法在更多的靶标上成功进行虚拟筛选;而相似分子对接方法之间,虚拟筛选性能互补性较差,比如HYBRID-M几乎全面优于FRED,没有体现出性能上的互补性。
总的来说,HYBRID比之FRED仅仅多花了一点CPU算力用于分子相似性比较,但是却极大地提高了分子对接虚拟筛选的早期富集能力。因此推荐在有holo结构可使用的条件下,尽可能多的使用多个holo结构用HYBRID进行虚拟筛选可以获得更好的早期富集能力。
5. 附件
5.1 六种分子对接方法的logAUC比较
5.2 GigaDock与GigaDock Wrap介绍
6. 文献
- OEDocking. https://www.eyesopen.com/oedocking
- McGann, M. R.; Almond, H. R.; Nicholls, A.; Grant, J. A.; Brown, F. K. Gaussian Docking Functions. Biopolymers 2003, 68 (1), 76–90. https://doi.org/10.1002/bip.10207.
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51 (3), 578–596. https://doi.org/10.1021/ci100436p.
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided. Mol. Des. 2012, 26 (8), 897–906. https://doi.org/10.1007/s10822-012-9584-8.
- Cleves, A. E.; Jain, A. N. Structure- and Ligand-Based Virtual Screening on DUD-E + : Performance Dependence on Approximations to the Binding Pocket. J. Chem. Inf. Model. 2020, 60 (9), 4296–4310. https://doi.org/10.1021/acs.jcim.0c00115.
- Spruce. https://www.eyesopen.com/spruce
- DUDE. http://dude.docking.org
- OMEGA. https://www.eyesopen.com/omega
- 肖高铿. 如何进行虚拟筛选的方法学验证. 墨灵格的博客. 2016-09-22. http://blog.molcalx.com.cn/2016/09/22/virtual-screening-methodology-validation.html
- Shen, C.; Hu, Y.; Wang, Z.; Zhang, X.; Pang, J.; Wang, G.; Zhong, H.; Xu, L.; Cao, D.; Hou, T. Beware of the Generic Machine Learning-Based Scoring Functions in Structure-Based Virtual Screening. Brief. Bioinform. 2021, 22 (3), 1–22. https://doi.org/10.1093/bib/bbaa070.
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61 (8), 3891–3898. https://doi.org/10.1021/acs.jcim.1c00203.
- Mysinger, M. M.; Carchia, M.; Irwin, J. J.; Shoichet, B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55 (14), 6582–6594. https://doi.org/10.1021/jm300687e.
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of Four Popular Virtual Screening Programs: Construction of the Active/Decoy Dataset Remains a Major Determinant of Measured Performance. J. Cheminform. 2016, 8 (1), 56. https://doi.org/10.1186/s13321-016-0167-x.
- Svensson, F.; Karlén, A.; Sköld, C. Virtual Screening Data Fusion Using Both Structure- and Ligand-Based Methods. J. Chem. Inf. Model. 2012, 52 (1), 225–232. https://doi.org/10.1021/ci2004835.
- 肖高铿. 用多个蛋白信息来提高用单一蛋白结构分子对接虚拟筛选的性能. 墨灵格的博客. 2022-03-02. http://blog.molcalx.com.cn/2022/03/02/oedocking-pde5a.html.


